Unusual Manifestations of Orbital Langerhans Cell Histiocytosis: Conjunctival Hyperemia and Ocular Hypertonia
Author: Lina Boualila1*, Adam Tagmouti1, Basma Mrini1, Nouredine Boutimzine1, Lalla Ouafae Cherkaoui1, Hafsa Elouazzani2, Nadia Cherradi2, Rania Bouanane3, Hamza Sbai3, Firdaous Touarsa3
1Department of Ophthalmology "A", Hospital of Specialities, Mohammed V University, Rabat
2Anatomopathology laboratory, Hospital of Specialities, Mohammed V University, Rabat
3Department of Radiology,Hospital of Specialities, Mohammed V University, Rabat
*Correspondence to: Lina Boualila, Department of Ophthalmology "A", Speciality Hospital, Ibn Sina University Hospital Center, Med V University, Rabat; E-mail: l.boualila@um5s.net.ma
Received: February 20, 2021; Accepted: February 26, 2022; Published: February 28, 2021
Citation: Boualila L, Tagmouti A, Mrini B, Boutimzine N and Cherkaoui LO (2022) Unusual Manifestations of Orbital Langerhans Cell Histiocytosis: Conjunctival Hyperemia and Ocular Hypertonia, 21st Century Pathol, Volume 2 (1): 112
Abstract
Langerhans cell histiocytosis (LCH) is a rare disease defined by the pathological proliferation of the monocyte-macrophage lineage and dendritic cells. Orbital involvement typically manifests as a solitary lesion that carries a favorable prognosis. We describe the clinical and histological profile of an orbital LCH case. The patient exhibited unifocal unisystem disease. Typical histologic features included numerous histiocytes with varying degrees of giant cell formation and scattered eosinophilic granulocytes. The presence of Langerhans cells was confirmed by CD1a and CD68 immunohistochemistry. We reviewed the different ophthalmic manifestations of LCH and treatment strategies. As LCH may solely involve the orbit, treatment is based on the degree of organ involvement. LCH should be included in the differential diagnosis of tumors of the ocular adnexa, especially in young children.
Keywords:
Langerhans cell histiocytosis; Rare disease; Proliferation; Monocyte macrophage lineage; Dendritic cells
Introduction
Langerhans Cell Histiocytosis (LCH) is a rare disease defined by the pathological proliferation of the monocyte-macrophage lineage and dendritic cells. It has long been referred to as histiocytosis X, a term covering Eosinophilic granuloma, Hand – Schuller – Christian disease, and Letterer –Siwe disease [1]. It can affect one or several organs. The orbital location of LCH represents 1% of orbital tumors [2]. It is often a uni-focal uni-system disease but can evolve to a multi-system disease. The symptomatology is a large mosaic, ranging from discrete to patent. The diagnosis is based on histopathological analysis. The extension assessment is an important step in medical care, it conditions therapeutic management and prognosis. Even if the therapeutic arsenal is varied, there is no standardized protocol. Many authors have reported successful outcomes after biopsy, curettage, and intralesional corticosteroid injection [3-6]. The present study aims to describe the atypical clinical presentation of LCH of the orbit and the different therapeutic strategies.
Clinical Observation
A 15-year-old child, with no particular medical history, was admitted to the emergency room for acute redness and pain of the left eye. The ophthalmological examination found (Figure 1: A, B, C, D): A visual acuity of 20/20 in both eyes, intraocular pressure at 35 mmHg in the right eye and 30 mmHg in the left eye. On the right eye, we found discreet upper eyelid edema, conjunctival hyperemia localized in the external canthus, with a bluish appearance of the episclera. The rest of the examination found no other abnormality.
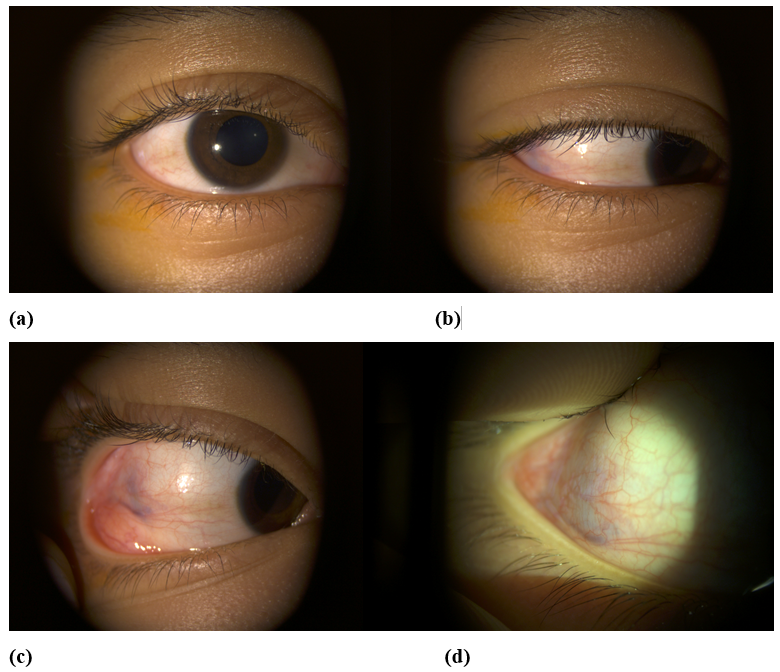
Figure 1: Pictures of the right eye (a), in primary position (b), (c) sideway gaze showing conjunctival hyperemia localized in the external canthus with a bluish appearance of the episclera(x 6,3), (d) microscopic magnification (x 16).
An inflammatory blood test has been requested: hemogram, reticulocytes, blood smear, sedimentation rate, and C reactive protein, with no anomaly found. Orbito-cerebral CT scan was performed, which showed an extra conical nodular thickening centered on the lateral rectus of the right orbit, associated with sphenoid lysis (Figure 2). 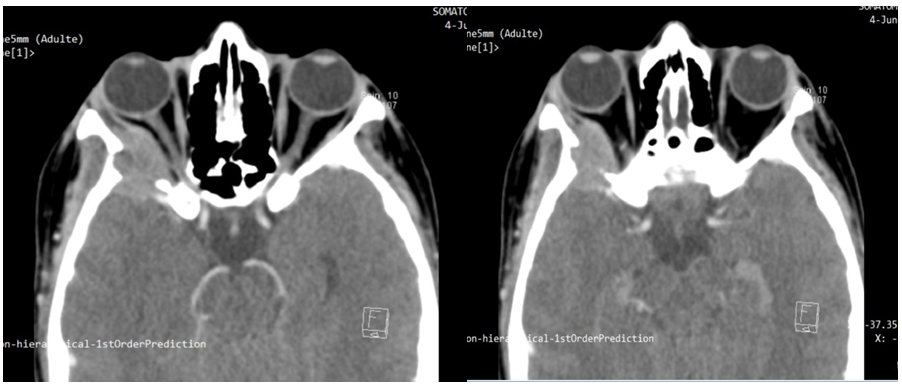
Figure 2: Cross-sections of orbital-cerebral CT showing an extra conical nodular thickening centred on the lateral rectus of the right orbit, associated with sphenoid lysis.
On MRI, the nodular lesion is hyposignal T2, isosignal T1, diffusion hypersignal, and intensely enhanced after injection. Pachymeningeal contrast on the endocranial side is related to reactive pachymeningitis (Figure3). 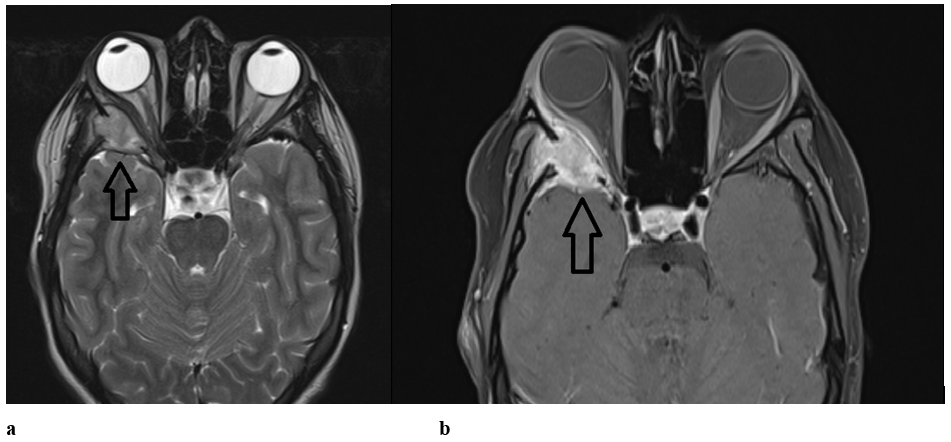
Figure 3: Axial sections of brain MRI (arrow) showing a nodular lesion hyposignal T2 and isosignal T1 (a) and intensely enhanced after injection (b).
A treatment with beta-blocker eye drops and oral antalgic has been initiated. Afterward, a lateral orbitotomy biopsy was performed to make a definitive diagnosis. The morphological and immunohistochemical study showed positivity for anti CD1a and anti CD68 at the histiocytic level (Figure 4). The diagnosis was in favor of Langerhansian histiocytosis.
An extension assessment was conducted, finding no other localization of the LCH. The post-biopsy evolution was marked by the spontaneous normalization of the intraocular and the disappearance of pain. A monthly check-up was done by measuring tone, visual field, and papillary OCT. 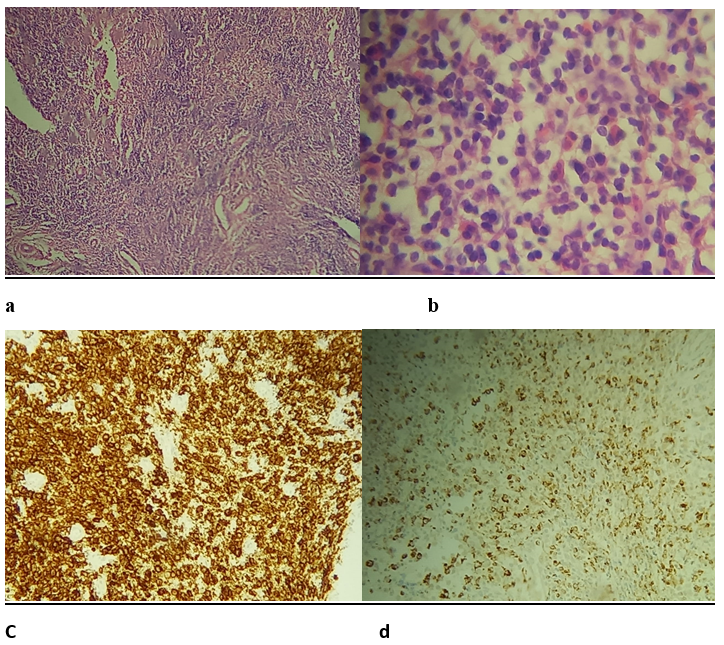
Figure 4: Microphotograph (hematoxylin and eosin stain, (a) 10 × and (b) 40×) showing numerous multinucleated giant cells, Langerhans cells with characteristic membrane grooving of the eosinophils, and showing diffuse immunoreactivity for CD1a protein and CD68 (c and d).
COVID-19 association with the complement system
Eosinophilic granuloma, Hand – Schuller – Christian disease, and Letterer- Siwe disease used to be referred to as Histiocytosis X. This was the case until the revision of the Histiocytic Society, defining Langerhans cell histiocytosis as the pathological proliferation of the monocyte-macrophage lineage and dendritic cells [1].
Langerhans histiocytosis boasts impressive flexibility in its affection, being able to affect an organ to multiple ones, and be uni or multifocal at their level. Its targets mainly comprise skin, bones, liver, spleen, lung, and central nervous system [2].
The most common ophthalmologic site is the rim of the frontal bone and the superior orbital roof, often in the form of isolated bone lysis associated with an endo-orbital mass [7]. Although the pathogenesis of Langerhans cell histiocytosis is not entirely understood [8], the frequency of its ophthalmic location may be explained by the presence of active hematopoietic marrow of the frontal bone’s medullary space. With age, the frontal sinus dilates and the active hematopoietic zone is limited to the lateral orbital roof, which aligns with the very frequent superolateral roof location after the age of 8 years [9]. In some cases, such as our patient, hematopoietic activity in the sphenoid's greater wing may persist up to the second decade of life, which may explain that location in young adults [7, 9].
Statistically, orbital involvement represents 1-20% of unifocal-single system involvement, and globally constitutes 1% of orbital tumors [2]. Some European studies find an incidence rate varying between 4 and 6 / 1,000,000 children per year, with an incidence of 9 to 15.3 / 1,000,000 per year for children under the age of 1, and 0.7 to 2 / 1,000,000 per year for those over 10 years old [10-13]. Although the diagnosis can be made at any age, two peaks of frequency stand out: between 1 and 4 years old, and in the third decade of life [14, 15].
From a clinical point of view, presentation depends on the tumor location and size. Anterior orbital location often results in ptosis and a mass or edema of the eyelid that can be mistaken for an infectious process. Posterior orbital location on the other hand may come with a proptosis, generally marked, along with diplopia, secondary to muscle infiltration or strabismus. Other rare locations have been reported, involving the eyelid, conjunctiva, caruncle, epibulbar nodule, choroid, optic chiasm, orbital apex, and cavernous sinus [13]. Visual acuity is usually preserved, with the exception of cases with papillary or macular edema.
The International Histiocytic Society has established criteria for Langerhans Cell Histiocytosis diagnosis. At least two items from the following are necessary: Positive staining for Adenosine triphosphate, S-100 protein, Alpha mannosidase, Peanut lectin binding. Definitive diagnosis requires the demonstration of Birbeck granules by electron microscopy or CD1a positivity [18]. Birbeck granules are rod-shaped, pentalaminar structures with vesicular end with an antigen presentation role. They are considered pathognomonic of LCH if present in 50-70% of the lesions [13].
Once the diagnosis of certainty is made, investigations to detect other possible locations are necessary. This is an important step, conditioning the treatment and the prognosis. Systemic evaluation should include complete hemogram, liver function test, coagulation profile, urine analysis and osmolarity, water deprivation test, chest X-ray, bone marrow analysis, a complete skeletal survey X-ray or PET scan, and imaging of abdomen and pelvis [19].
The differential diagnoses can be divided into clinical and histological. The clinical differential diagnoses for children are lymphoma, Ewing's sarcoma, leukemia, metastases, dermoid cyst, orbital cellulitis, acute dacryocystitis, or bone infection such as bone tuberculosis. For adults, differentials mainly consist of lacrimal gland tumors, meningiomas, dermoid cysts, and metastases [9]. The histological differential diagnosis is giant cell reparative granuloma, hemorrhagic cyst, cholesterol granuloma, Erdheim-Chester disease, giant cell aneurysm of bone, giant cell tumor, and the rare histiocytic and Langerhans cell sarcomas [13].
Concerning the prognosis, unifocal lesions present a better outcome than multisystemic lesions. Endocranial extension of an orbital LCH, considered a unifocal lesion [20], is correlated with a risk of developing diabetes insipidus [13].
Based on a systematic review, Bezdjian A, et al. (2015) created an algorithm to resume the diagnosis steps and guide the treatment (Figure 5) [21].
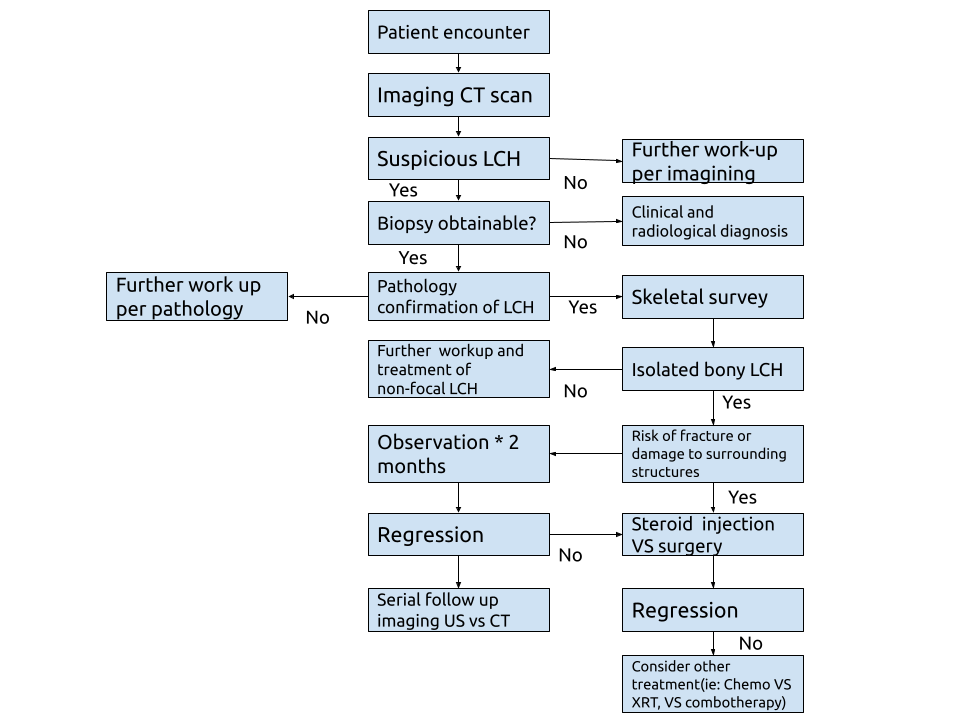
Figure 5: Treatment algorithm for the diagnosis and treatment of Langerhans cell histiocytosis (LCH) isolated bone lesions in the head and neck. CT, computed tomography; US, ultrasound.
A review of the literature shows the lack of standardized protocol for therapeutic management. Multiple factors must be taken into consideration: location of the lesion, uni or multifocality, whether it affects single or multiple organs, and recurrences. Many therapeutic approaches exist observation after biopsy, curettage, surgical excision, corticotherapy, radiotherapy, and chemotherapy.
For solitary orbital lesions, such as our case, biopsy and curettage are recommended by many authors. Spontaneous resolution following biopsy was reported by the likes of Smith JH, et al. (1999) and Glover AT, et al. (1987) [3, 4], while Rajendram J, et al. (2005) reported the resolution of the orbital LCH after curettage and intralesional steroids [5]. This mysterious outcome may be explained by the interruption of the pathological cascade through the modification of the microenvironment [6].
To prevent recurrences, intralesional corticotherapy post-excision at the dose of 125mg of methylprednisolone is recommended. It inhibits IL-1 and PGE2, and mediates osteolysis. Harris et al treated four out of seven patients with intralesional steroid post-excision and reported nil recurrence after a follow-up period of 6–24 months [6].
As for multifocal/multisystem disease or recurrences and in orbital lesions with dural involvement, chemotherapy constitutes a valid option [19, 22 and 23]. The LCH III protocol recommended systemic chemotherapy to prevent the development of “central nervous system” (CNS) complications, considering orbital disease as a CNS risk lesion [24]. Although there is no direct evidence proving the effect of chemotherapy in the prevention of progression to the central nervous system in the cases of unifocal orbital lesions [25].
Radiotherapy is usually proposed to treat recurrences [13]. Das et al documented regression without any recurrence at 4 years of follow-up in an 8-year-old girl with orbital LCH who received 1500 cGy in three fractions [23].
Last but not least, bone marrow transplantation and immunoglobulin therapy are reserved for uncontrolled disease recurrences and central nervous system involvement, respectively [26, 27]. Despite the Histiocyte Society presenting the latest treatment strategies, there is no standardized protocol for ophthalmological LCH. These trials are focused on multi-system disease and are not conducted by ophthalmologists [28, 29].
Conclusion
This case report highlights the importance of CT-scan in front of atypical inflammatory symptomatology with ocular hypertonia and the necessity to discuss tumor pathologies, especially in children.
Favipiravir is an antiviral prodrug that is converted to an active metabolite through phosphorylation by the intracellular enzyme hypoxanthine-guanine phosphoribosyl transferase (HGPRT). It shows non-CYPs facilitated biotransformation by aldehyde oxidase (AO) and xanthine oxidase (XO). Variants of AO are frequently linked to pharmacodynamics (PD) in other medications that are AO's substrate. Because of the genetic variability of AO, elevated plasma levels of favipiravir must be indicated in Asian populations. Based on AO, the DDI of zaleplon and cimetidine is already known. Cimetidine co-administration inhibits AO-catalyzed oxo-zaleplon formation, and the Zaleplon level is cautiously included. Clinicians should be aware that co-administration of the medicine with an AO inhibitor, such as cimetidine, tamoxifen phenothiazines, verapamil, amlodipine, nifedipine, loratadine, cyclobenzaprine, ondansetron, or ketoconazole, could theoretically increase plasma levels of favipiravir active metabolites because of possible drug-drug interactions. Its metabolite inhibits OAT1 and OAT3 in a moderate way [141]. It is also a moderate inhibitor of various CYPs, but its effect on CYP2C8 must be clinically significant to enhance exposure [187] to drugs like rosiglitazone (hypoglycemic), repaglinide, torasemide, paclitaxel, and buprenorphine.
The hydroxychloroquine (HCQ) metabolism is hepatic but not been precisely characterized and the effect CYP2C8 CYP2D6 and CYP3A4/5 being extrapolated from chloroquine (CQ) data. The half-life of HCQ elimination is approximate 40 days. Both CQ and HCQ transform into active metabolites by CYP isomers through the dealkylation process [213, 214].
Systemic Lupus Erythematosus (SLE) patients demonstrated that HCQ and CQ both have variability in metabolism and the effect of CYP2D6 SNPs on blood HCQ level [215]. The gene polymorphisms of CYP2C8 CYP2D6 and CYP3A4/5 may also alter the disposition. As immunologically challenged patients frequently developed more severe COVID-19 clinical complications. HCQ medication treatment for COVID-19 infection was contested by various side effects among individual variability in CYP genotypes. Specifically, CYP2D6 genotyping may be helpful to decide the best possible HCQ dosage in the context of personalized medicine. CQ and HCQ are both inhibitors of P-gp. Both drugs also increase cyclosporine levels, with HCQ increasing digoxin levels. The multiple DDI that are now known, as well as the potential use of these agents in combination with other drug therapies, requires consideration for the safety of the patients.
Azithromycin is an antibiotic belonging to the macrolide group, used to prescribe COVID-19 patients who developed the risk of secondary infection. It has high tissue affinity with wide distribution in the body. The elimination half-life of azithromycin is between 2-4 days. It is primarily eliminated via the liver. It is noticed mainly in the bile and urine as a parental form. It inhibits CYP3A4 [198], OATP1A2 and OATP2B1 [173] moderately. Azithromycin and HCQ both drugs are both metabolized by CYP3A4. This combination was revealed to decrease the mortality associated with COVID-19 infection [216]. Co-medication of azithromycin heightened the QT of HCQ results enhance the chances of cardiac failure and cardiovascular mortality [217]. The possibility of a probable interaction is not PK but PD while recommending azithromycin as co-medication in connection with its effect on QT prolongation. Risks should be evaluated when azithromycin is administered to COVID patients with impaired hepatic function.
Dexamethasone is a synthetic glucocorticoid. It is 6-hydroxylated (6α- and 6β-hydroxy dexamethasone) by CYP3A4 and reversibly metabolized to dexamethasone by 11β- dehydrogenase isozyme. Dexamethasone is an agonist of nuclear pregnane X receptor (PXR) [218] and It is a weak inducer of CYP3A4 [219, 220] through PXR, with most data indicating that it decreases the exposure of sensitive CYP3A4 substrates by approximately 20%. PXR also changed the expression of other drug-metabolizing enzymes and transporters, such as CYP3A11, CYP2B10, and OATP2 [221-223]. Drug metabolizing enzyme variants (CYP3A4, CYP3A5, CYP3A7, and GSTT1) and transporters (ABCB1 and MDR1) have been associated with response to corticosteroids in several diseases [224]. Overdoses of corticosteroids are considered sufficiently immunosuppressive to warrant unease about probably lessening the effectiveness of vaccines. Corticosteroids also have a high risk of producing or aggravating hyperglycemia and may diminish the efficacy of antidiabetic drugs.
Ivermectin is presented as necessary medicine of the WHO model list. Ivermectin is mostly metabolized by CYP3A4, and it inhibits CYPC9, CYP2C19, CYP2D6, and CYP3A4 [197]. In animals, Ivermectin DDIs begin mainly at the level of P-gp (ABCB1). It is a substrate for P-gp [225] that enables ivermectin intestinal and biliary excretion and prevents it from entering the CNS (Central Nervous System) [226, 227]. Other transporters might also be involved in ivermectin DDIs like MRPs and BCRP [228]. Pgp and CYP3A4 inhibitors may increase the ivermectin level in the plasma. Ideally, metabolism might decrease with age leading to higher exposure of ivermectin in aged patients [229].
Umifenovir is also described in the publication as Arbidol. It was mainly excreted in urine as phase II conjugates with glucuronide and glucuronide sulfinyl as major metabolites. Arbidol is a substrate for CYP3A4 and FMOs in vitro, insinuating that use with CYP3A4 inducer and inhibitors may lead to possibly significant increases and decreases in arbidol exposure. Total 33 arbidol metabolites were found in human plasma, urine, and feces by engaging the different CYPs including CYP1A1, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, FMO1, FMO3, and FMO5 [194]. Three major metabolites (M5, M6-1, and M8) were detected in the plasma following oral treatment of umifenovir to healthy individuals. M6-1 is critical to assess for safety and efficacy because of its high exposure and extended elimination half-life. CYP3A4 was the most active enzyme in the arbidol metabolism in the liver and gut, followed by other CYPs and FMOs [194]. However, concern and close monitoring are advisable when it is on medication, and medical practitioners are reassured to inform presumed DDI that they observe concerning arbidol.
Ruxolitinib and baricitinib, the Janus kinase inhibitor that has been used to treat COVID-19. Ruxolitinib is mostly metabolized by CYP3A4 and CYP2C9, with CYP1A2 and CYP2B6 playing a minor role [200, 201]. Following inhibition of both CYP2C9 and CYP3A4 enzymes, fluconazole may increase plasma exposure. On the basis of the PBPK model, the importance of this interaction was validated in healthy subjects [230]. Fluconazole simulation findings were used as a foundation for ruxolitinib dose correction when perpetrator drugs were co-administered. Baricitinib clinical drug-drug interaction still needs to be known. It is metabolized in part by CYP3A4 and is a substrate of P-gp, BCRP, OAT3, multidrug, and toxin extrusion protein (MATE) 2K. OCT1 is likewise inhibited by baricitinib. Probenecid is used for treating gout disease, increasing the exposure of baricitinib following inhibition of OAT3, and lowering the renal clearance in healthy participants. The renal clearance and inhibitory effect of probenecid on baricitinib were reproduced in vitro using PBPK modelling [180]. Both have immunosuppressive effects and associated interactions concerns regarding other immunosuppressants and live vaccines.
Imatinib is the tyrosine kinase inhibitor and is solely metabolized by CYP3A4 and CYP2C8 [204]. It was also shown that CYP3A4 inhibition occurs through an irreversible mechanism [231]. The time-dependent auto inhibition of its own CYP3A4 metabolism leads to an important role for CYP2C8 in the elimination of imatinib. The auto inhibition of its own CYP3A4 metabolism in a time-dependent manner resulted in a significant involvement for CYP2C8 in imatinib elimination. Interactions with other drugs and pharmacogenetic polymorphisms both impact CYP2C8 activity during many administrations, resulting in apparent interindividual variability in imatinib exposure. Dexamethasone is a potent inducer of CYP3A4 and significantly reduces the plasma level of imatinib. However, there is no literature available to find DDI with imatinib and dexamethasone. Imatinib is a known substrate of ABC transporters [232, 233], they have been reported to antagonize these transporters. This effect could manipulate the PK properties of imatinib, perhaps lessening its absorption subsequently causing imatinib concentration to drop below therapeutic levels [234, 235].
Fluvoxamine is a serotonin reuptake inhibitor with a half-life of approximately 30 hours. It has anti-inflammatory properties. It is known that it inhibits the enzyme CYP1A2. It is, however, a strong inhibitor of CYP2C19. A case study of a 48-year-old lady with a psychiatric disorder has died following treatment of multiple drugs along with fluvoxamine. After the autopsy, femoral and cardiac blood, urine, and bile were collected for toxicological kinetics. Fluvoxamine, propranolol, clotipine, gabapentin, 7-aminoclonazepam, and haloperidol drugs were detected after the examination. Fluvoxamine levels in the blood exceeded the upper limit of therapeutic blood level by approximately ten times. The relevant cause of death was due to multiple drug use [236]. Fluvoxamine has a minor effect on medications that are metabolized by CYP3A4. It is classified as a potent inhibitor of CYP1A2 and CYP2C19 by the FDA. It is commonly used for clinical DDI [237]. PBPK model was created to characterize the CYP1A2 and CYP2C19 for interaction between fluvoxamine and other drugs [238]. As a result, it can be used to support prospective dose adaptation suggestions, labelling, and clinical DDI trial design.
Sofosbuvir is a substrate of Pgp and BCRP [185]. Hydrolases metabolize sofosbuvir extensively, and GS-331007 metabolite accounts for more than 90 % of overall drug exposure [209]. The metabolizing enzymes and drug transporters are not inhibited or induced by sofosbuvir or its metabolites. Daclatasvir is a substrate for CYP3A4 Pgp and also the inhibitor of Pgp, BCRP, OATP1B1, and OATP1B3 [186]. The feces are the predominant route of elimination for daclatasvir, followed by the kidneys. Sofosbuvir and daclatasvir have exhibited good safety profiles with minimum drug interaction and widely available therapeutic options for COVID-19 treatment.
Some of the drugs included in Table 1 lack pharmacogenomic data and are still in clinical trials for the treatment of COVID-19 infection, therefore more research is needed to determine drug metabolism and disposition in the inflammatory and immunomodulatory state.
Conclusion
COVID-19 is classified as a multisystemic disease. The elementary pathogenesis involves distinct components; immune deficiency and severe lung inflammation, which leads to increased production of cytokines and is related to inappropriate immune response. Timely management of the cytokine storm in its early stage through immunomodulators, cytokine antagonists as well as the reduction of lung inflammatory cell infiltration, are the key to reducing the mortality rate and improving the treatment success rate of COVID-19 patients. In consequence, treatment approaches at present include anti-infectious, anti-viral, anti-proinflammatory cytokines, and life support therapies. The potential of disease-drug or drug-drug interaction is an essential concern while providing optimal treatment regimens for individual patients. Hence, the present review expounded the probability of drug interaction in COVID-19 patients while reaching inflammatory conditions and or having comorbidity like autoimmune and metabolic diseases. This point is highly clinical relevance for drugs with a narrow therapeutic index like HCQ and CQ. As the mechanism underlying the observed alteration in the plasma binding proteins, drug transporters, and DMEs comes to be clear, the next step is to address the clinical importance to better predict the drug concentration in the inflammatory state of the disease condition. Proinflammatory cytokines impede the expression and activity of drug transporters and DMEs (hepatic and extrahepatic). Alteration in the transporters and DMEs can lead to changes in the PK parameters of used drugs. The risk of drug interactions should not be prohibited since they are frequently manageable and convenient. The consumption of a single drug may possibly not be more effective, but during co-medication of multiple drugs, the risk of drug interaction must be increased. The impact of inflammation on the PK of the drug remains and alternated an important field of research.
Funding
Authors have not received any funding from any private and government organization.
Conflict of Interest
The author have no conflict of interest.
Overlapping Publication
This article have not been submitted to any other journal for publication.
References
1. Margo CE, Goldman DR (2008) Langerhans cell histiocytosis. Survey of Ophthalmol 53: 332-358. https://doi.org/10.1016/j.survophthal.2008.04.007
2. Broadbent V, Gadner H (1998) Current therapy for langerhans cell histiocytosis. Hematology/Oncology clinics of North America 12: 327-338. https://doi.org/10.1016/s0889-8588(05)70513-x
3. Smith JH, Fulton L, O’Brien JM (1999) Spontaneous regression of orbital Langerhans cell granulomatosis in a three-year-old girl. Am J Ophthalmol 128: 119-121. https://doi.org/10.1016/S0002-9394(99)00055-0
4. Glover AT, Grove AS Jr (1987) Eosinophilic granuloma of the orbit with spontaneous healing. Ophthalmol 94: 1008-1012. https://doi.org/10.1016/s0161-6420(87)33352-4
5. Rajendram R, Rose G, Luthert P, et al. (2005) Biopsy-confirmed spontaneous resolution of orbital langerhans cell histiocytosis. Orbit 24: 39-41. https://doi.org/10.1080/01676830590889893
6. Harris GJ, Woo KI (2003) Eosinophilic granuloma of the orbit: a paradox of aggressive destruction responsive to minimal intervention. Trans Am Ophthalmol Soc 101: 93-103, 103-105.
7. Cheung N, Selva D, McNab AA (2007) Orbital langerhans cell histiocytosis in adults. Ophthalmology 114: 1569-1573. https://doi.org/10.1016/j.ophtha.2006.10.056
8. Favara BE (2001) Langerhans cell histiocytosis: an identity crisis. Med Pediatr Oncol 37: 545. https://doi.org/10.1002/mpo.1250
9. Koka K, Alam MS, Subramanian N, et al. (2020) Clinical spectrum and management outcomes of Langerhans cell histiocytosis of the orbit. Indian J Ophthalmol 68: 1604-1608.
10. Salotti J (2008) Epidemiology of Langerhans cell histiocytosis: onwards and upwards! Pediatr Blood Cancer 51: 3-4. https://doi.org/10.1002/pbc.21552
11. Guyot-Goubin A, Donadieu J, Barkaoui M, et al. (2008) Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000-2004. Pediatr Blood Cancer 51: 71-75. https://doi.org/10.1002/pbc.21498
12. Alston RD, Tatevossian RG, McNally RJQ, et al. (2007) Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer 48: 555-560. https://doi.org/10.1002/pbc.20884
13. Herwig MC, Wojno T, Zhang Q, et al. (2013) Langerhans cell histiocytosis of the orbit: five clinicopathologic cases and review of the literature. Surv Ophthalmol 58: 330-340. https://doi.org/10.1016/j.survophthal.2012.09.004
14. Hamre M, Hedberg J, Buckley J, et al. (1997) Langerhans cell histiocytosis: an exploratory epidemiologic study of 177 cases. Med Pediatr Oncol 28: 92-97.
15. Aricò M, Girschikofsky M, Généreau T, et al. (2003) Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer 39: 2341-2348. https://doi.org/10.1016/s0959-8049(03)00672-5
16. Gross FJ, Waxman JS, Rosenblatt MA, et al. (1989) Eosinophilic granuloma of the cavernous sinus and orbital apex in an HIV-positive patient. Ophthalmol 96: 462-467.
17. Gündüz K, Palamar M, Parmak N, et al. (2007) Eosinophilic granuloma of the orbit: report of two cases. J AAPOS 11: 506-508.
18. Malone M (1992) The histiocytoses of childhood. Histopathol 21: 395-396. https://doi.org/10.1111/j.1365-2559.1992.tb00419.x
19. Kramer TR, Noecker RJ, Miller JM, et al. (1997) Langerhans cell histiocytosis with orbital involvement. Am J Ophthalmol 124: 814-824. https://doi.org/10.1016/s0002-9394(14)71699-x
20. Shetty SB, Mehta C (200) Langerhans cell histiocytosis of the orbit. Indian J Ophthalmol 49: 267-268.
21. Bezdjian A, Alarfaj AA, Varma N, et al. (2015) Isolated langerhans cell histiocytosis bone lesion in pediatric patients: systematic review and treatment algorithm. Otolaryngol Head Neck Surg 153: 751-757. https://doi.org/10.1177/0194599815598969
22. Holbach LM, Colombo F, Heckmann JG, et al. (2000) Langerhans-cell histiocytosis of the orbit; diagnosis, treatment and outcome in three patients -- children and adults. Klin Monbl Augenheilkd 217: 370-373. https://doi.org/10.1055/s-2000-9578
23. Das JK, Soibam R, Tiwary BK, et al. (2009) Orbital manifestations of langerhans cell histiocytosis: A report of three cases. Oman J Ophthalmol 2: 137-140. https://doi.org/10.4103/0974-620x.57315
24. LCH - III 2nd version (2002) Treatment protocol of the third international study for langerhanscell histiocytosis. https://www.skion.nl/workspace/uploads/lchiiiprot-version2.pdf
25. Esmaili N, Harris GJ (2016) Langerhans cell histiocytosis of the orbit: spectrum of disease and risk of central nervous system sequelae in unifocal cases. Ophthal Plast Reconstr Surg 32: 28-34. https://doi.org/10.1097/iop.0000000000000402
26. Gavhed D, Laurencikas E, Akefeldt SO, et al. (2011) Fifteen years of treatment with intravenous immunoglobulin in central nervous system Langerhans cell histiocytosis. Acta Paediatr 100: e36-39. https://doi.org/10.1111/j.1651-2227.2010.02125.x
27. Conter V, Reciputo A, Arrigo C, et al. (1996) Bone marrow transplantation for refractory Langerhans’ cell histiocytosis. Haematologica 81: 468-471. https://doi.org/10.3324/%25x
28. Minkov M (2011) Multisystem langerhans cell histiocytosis in children: current treatment and future directions. Paediatr Drugs 13: 75-86. https://doi.org/10.2165/11538540-000000000-00000
29. Allen CE, McClain KL (2007) Langerhans cell histiocytosis: a review of past, current and future therapies. Drugs Today 43: 627-643. https://doi.org/10.1358/dot.2007.43.9.1088823