Immunomodulation may Affect the Efficacy of COVID-19 Medication by Changing Drug Metabolism and Pharmacokinetics
Author: Devendra Kumar1*, Neerja Trivedi2, Arti Verma3
1Department of Pharmaceutical Sciences, University of Nebraska Medical Center, Omaha, NE, USA
2Department of Pharmacology and Neuroscience, School of Medicine, Creighton University, Omaha, NE, United States.
3Clinical and Experimental Therapeutics, College of Pharmacy, University of Georgia, and Charlie Norwood VA Medical Center, Augusta, GA, USA
*Correspondence to: Devendra Kumar, Department of Pharmaceutical Sciences, University of Nebraska Medical Center, Omaha, NE, USA; E-mail: devendra.kumar@unmc.edu
Received: February 17, 2021; Accepted: February 26, 2022; Published: February 28, 2021
Citation: Devendra Kumar, Neerja Trivedi and Arti Verma (2022) Immunomodulation may Affect the Efficacy of COVID-19 Medication by Changing Drug Metabolism and Pharmacokinetics, 21st Century Pathol, Volume 2 (1): 111
Abstract
The COVID-19 outbreak, which was prompted by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus, continues to spread over the world. In the pathophysiology of COVID-19, the immunological response of the patient is crucial. The immunological changes occur when the immune system is fighting against SARS-CoV-2 and the signal becomes hyperactive. In drug metabolism and transport mechanism, a hyperimmune state can cause severe abnormalities. As a result, inter-individual variability in drug pharmacokinetics may occur, potentially leading to unexpected treatment responses. The physiology of drug transporters and drug-metabolizing enzymes are also affected in COVID-19 patients with autoimmune and metabolic disorders. The Food and Drug Administration (FDA) has approved several antiviral and anti-inflammatory drugs to treat COVID-19 patients based on the evidence, despite the fact without knowing any possible interactions with each other. Inflammation directs the altered expression of drug-metabolizing enzymes and drug transporters, shown by preclinical and clinical research. A deeper understanding of the impact of immune responses on drug metabolism and transport, as well as the therapeutic implications of this knowledge, would aid in the personalization of drug treatment. The current review summarizes the effect of inflammation and the complement system on the pharmacokinetics of COVID-19 medications by altering the expression and activity of drug transporters and drug-metabolizing enzymes, as well as offers an opinion on possible drug-drug interactions in disease states. More studies, including control PK trials, are needed to determine the clinical significance in the COVID-19 infected population. The results of future studies will facilitate us to better predict concentrations in individuals who have hyperimmune responses so that we can provide them personalized medicine.
Keywords:
COVID-19; Inflammation; IL-6; Complement Systems; Drug transporters; CYP450
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-Co-2) infection emerged as a pandemic in early 2020 year and is commonly known as COVID-19 [1]. The first case of COVID-19 in pneumonia patients was detected in Wuhan city, China in December 2019. In the beginning, pneumonia patients showed normal respiratory infection, which rapidly transformed into acute respiratory syndrome [2]. Acute respiratory syndrome was associated with chronic inflammation. Chronic inflammation occurs when an antigen is constantly present in the body, and the immune system is continuously activated to remove the infection. This constant inflammation causes some degree of alteration in the drug-binding proteins [3-5]. Recent studies have been suggested that inflammation can modulate transporters and drug-metabolizing enzymes (TDMEs) [6]. The downstream regulation of various hepatic and extrahepatic drug-metabolizing enzymes (DMEs) was already shown by some studies [7, 8]. Inflammation-induced alterations in the expression of a variety of membrane-associated drug transporters have recently been described [9]. Therefore, a better understanding of the impact of inflammation on TDMEs and its associated clinical outcomes would help to recognize the individual pharmacokinetic (PK) and pharmacodynamic (PD) variability of drugs. The SARS-CoV-2 infection resulted in increased frequency of hospitalization and intensive care unit (ICU) admission [9]. This is considered a major clinical concern that critically ill patients are more affected by drug interaction that TDMEs are involved in the transport and metabolism of commonly prescribed drugs [10]. The high prevalence of acute and chronic inflammatory conditions, the modulation in the expression and activity of TDMEs may modify the PK/PD of drugs used in COVID-19 treatment (Figure 1).
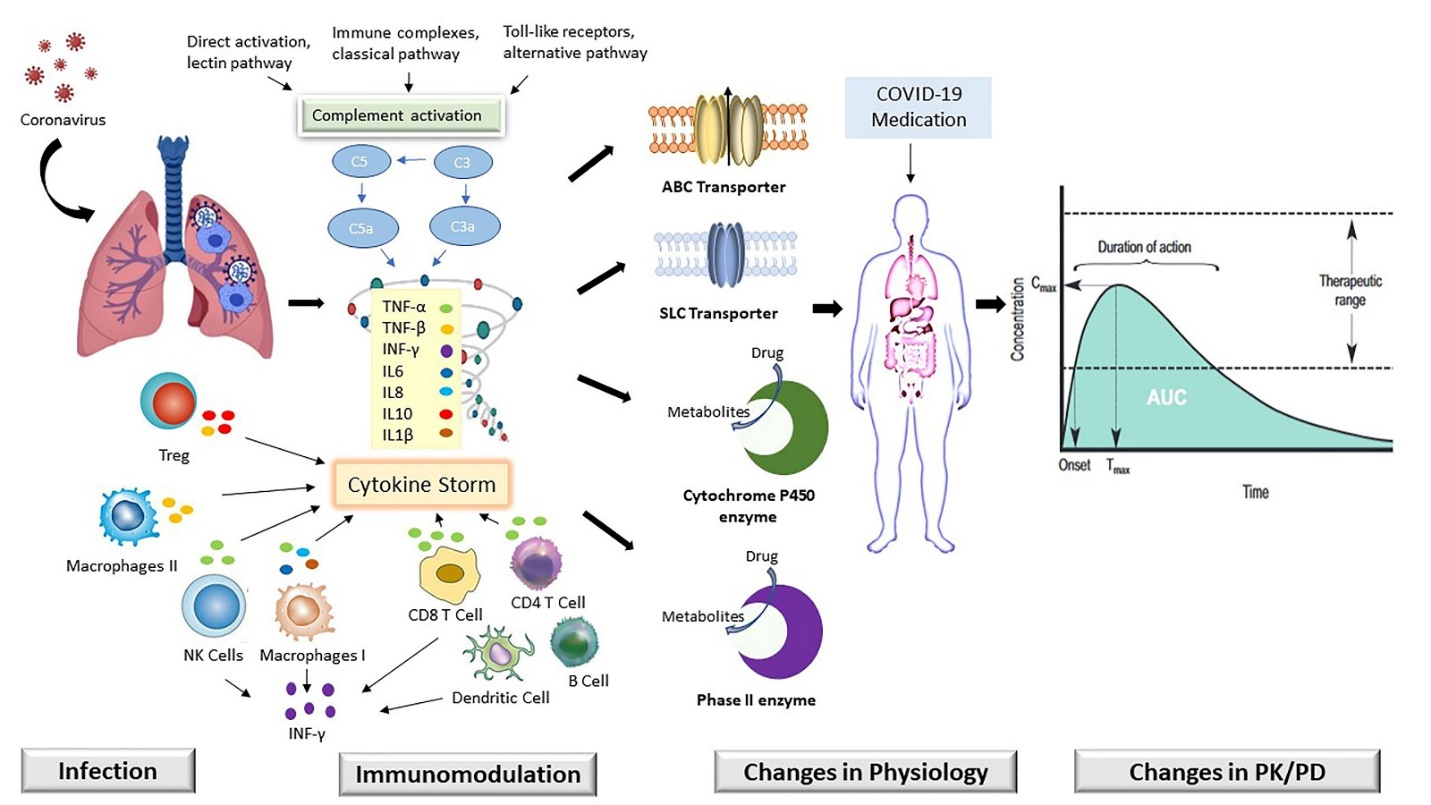
Figure 1: Modification in the expression of key transporters (ABC: ATP binding cassette and SLC: Solute carrier) and drug metabolizing enzyme (Cytochrome P450 and Phase II) mediated by acute immune response in SARC-CoV-2 (COVID-19) infection play a role in alteration of drug absorption distribution metabolism and excretion (ADME).
Based on the evidence, the Food and Drug Administration (FDA) approved some drugs that have been already used (Table 1) for the treatment of SARC-CoV-2 [11, 12]. In these drugs, remdesivir and favipiravir have been proven most promising against COVID-19 treatment. The lopinavir-ritonavir combination showed to be effective against COVID-19. Other antiretroviral drugs atazanavir and darunavir were also used. Anti-malarial drug hydroxychloroquine showed good results against COVID-19, and it has been used frequently in combination with other drugs [13], [14]. Irrespective of the antiviral drugs, dexamethasone showed little relief against COVID-19 but involved the increased risk of hyperglycemia. The initial clinical trial showed that Dexamethasone lowered the death of one-third of severe patients that were on a ventilator [15]. Sofosbuvir plus Daclatasvir in combination with Ribavirin was shown promising results to fight COVID-19 infection [16]. Fluvoxamine has also shown the potential in COVID-19 infected outpatients [17]. Molnupiravir, which can block the transmission of SARS-CoV-2 within 24 hours, is used to treat COVID-19 infection [18]. Some anti-inflammatory and anti-complement drugs were also used either in combination or alone for the treatment of COVID-19 patients.
This article will emphasize on the current state of knowledge and assume a PK and drug-drug interaction (DDI) potential of therapeutic agents used for co-morbidities and frequently used in intensive care for COVID-19 patients. Here, we present the subscription of inflammation on modulating the expression and activities of TDMEs and discuss its related clinical consequences of DDI based on drug metabolism and pharmacokinetics (DMPK) and finally provide the opinion on how to incorporate inflammation in pharmacological treatment.
Cytokine storm in COVID-19 patients
Cytokine storm is a complex and robust inflammatory phenomenon initiated by a group of cytokines mainly pro-inflammatory cytokines that are produced by the uncontrolled immune function. Adequate evidence showed very high levels of cytokines in severe COVID-19 patients. This cytokine storm is a range of cytokines, including interleukins (IL-1, IL-2, IL-6, IL-7, IL-8, IL-10, IL-12, IL-17, IL-18), tumor necrosis factor-α (TNF- α), interferon-γ (IFN- γ), granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF) and monocyte chemoattractant protein-1 (MCP-1) [19-27]. Cytokine targeted treatment in critical COVID-19 patents confirmed the presence of cytokine storm and showed the clinical advantages in hemophagocytosis [28, 29]. The COVID-19 cytokine storm has shown variability as normal cytokine storms in several aspects [19, 28, 30]. Cytokine storm in COVID-19 consists of more inflammatory cytokines as well as have an abnormally low level of lymphocytes (lymphopenia), suggesting that attributed to the innate immunity rather than adaptive immune cells [28]. This treatment approach is to manage ongoing inflammatory response by specifically or non-specifically targeting inflammatory cytokines or associated signaling pathways to resume the host immune regulatory system.
Immunomodulation in COVID-19
COVID-19 association with inflammation
Cytokines are the distinct class of small cell-signaling proteins responsible for maintaining the homeostasis of the immune system. Many studies have shown that, in addition to direct viral damage, unrestrained inflammation causes disease severity in COVID-19 patients [31, 32]. The high expression of inflammatory components, including C-reactive protein (CRP), ferritin, and increased levels of inflammatory cytokines and chemokines were noticed in COVID-19 infected patients [33-35]. One study was conducted to measure serum interleukin IL-6, IL-8, IL-1β, tumor necrosis factor (TNF) in hospitalized COVID-19 patients (n = 1484) for 41 days. Study results showed a high level of TNF-α, IL-6, and IL-8 at the time of hospitalization. Particularly, when adjusting for disease severity, IL-6 and TNF-α serum levels were constantly independent and significant predictors of severity and death [36]. Some proinflammatory cytokines such as interferon-gamma (IFN-γ), granulocyte-macrophage colony-stimulating factor (GM-CSF), macrophage colony-stimulating factor (MCSF), IL-12 were elevated in COVID-19 patients [2]. The cytokine profile in severe COVID-19 patients showed similarities like cytokine release syndromes, such as macrophage activation syndrome, with augmented production of cytokines and of chemokines including CCl-2 (CC-chemokine ligand -2) CCL3 and CXL10 (CXC-chemokine ligand -10) as well as the α-chain of the IL-2 receptor. This phenomenon has driven the hypothesis that dysregulated stimulation of the phagocytes is accountable to COVID-19 associated hyper inflammation [35, 37]. Inflammation is normal in reaction to damage or pathogenic illness, but it seems that patients with noncommunicable diseases reveal pre-heightened inflammatory levels. This can produce them subject to unrestrained inflammation when infected by the SARS-CoV-2, leading to a cytokine storm [38]. This is an excessive release of the cytokine in response to COVID-19 infection due to loss of regulation on the production of pro-inflammatory cytokines. Additionally, cytokine storm is anticipated to be the cause of acute respiratory distress syndrome (ARDS) and multiple organ failure [39, 40]. Obstructing these inflammatory elements can alter the developed immune response and stop lung destruction. IL-6 is the important target for anti-cytokine therapy and its elevation is mediated with a terrible diagnosis [29, 41, 42]. A monoclonal antibody against the IL-6 receptor named tocilizumab was used for the treatment of COVID-19 and demonstrated significant improvement in COVID-19 patients [43]. Another IL-6 receptor antagonist, sarilumab currently considered for COVID-19 treatment [44].
COVID-19 association with the complement system
The Complement system performs as immune surveillance and responds rapidly against infection. However, when dysregulation of these powerful machineries (C3a, C3b, iC3, C3c C3dg, C5), can become harmful and incriminate as a pathogenic effector in various diseases including viral infection [45]. Initially, it was reported that complement activation is attenuated by poxviruses by secreting a protein that showed structural and functional homology to the complement regulatory protein [46]. Flavivirus also hijacks the same and HIV-1 conscripts host complement system into their virion [45]. In all these scenarios, C3 attached to the surface of viruses and virus perform the cleavage of C3 that may affect the host antiviral response [47]. Whether SARS-CoV-2 exhibits such antiviral strategies remains to be answered.
COVID-19 mediated complement system activation
Promising, in-vitro, and in-vivo data analysis suggest that complement activation plays a crucial part in the pathogenesis and diseases seriousness of SARS-CoV and SARS-CoV-2. Gralinski LE, et al. (2018) found the effect of SARS-CoV infection using C3-/- mice and showed complement activation [48]. Mice after getting infected by SARS-CoV, in a 24-hour C3 activation product (C3a, C3b, iC3, C3c, and C3dg) were examined in the lungs. In the absence of C3, lung grievance and weight loss were significantly reduced, without altering the viral load, and cytokine and chemokine levels were found low as well in the C3 -/- mice. Moreover, factor B -/- or C4-/- mice also showed less weight loss [48]. These results showed that pulmonary pathology and SARS-CoV infection-associated illness can be improved by activating the complement system [49]. Nucleoprotein (secreted N protein) dimer of SARS-CoV-1, SARS-CoV-2 and MERS-CoV activate the MASP-2 enzyme (mannan-binding lectin-associated serine protease -2). This is a primary enzymatic inhibitor of the lectin pathway [50]. Auto activation of MASP-2 starts to generate C3 convertase and membrane attack complex (MAC). The interaction of MSP-2 and N protein attenuates lung injury. Human proteomic and metabolomics studies [51] along with these data suggest that coronavirus infection involves the activation of various pathways of the complement system.
In Italy, a single-center case series study showed that elevated level of plasma complements protein C5a and sC5b-9 were observed in patients with mild illness (patients requiring continuous positive airway pressure) and serious critical illness (mechanically ventilated) COVID-19 [52]. In COVID-19 patients, complement fragment deposition has been found in multiple organs. Extensive deposition of C5b-9, C4d, and MASP-2 in the microvasculature induced septal capillary damage in the lungs of COVID-19 patients who died because of respiratory failure [53]. Reports have been demonstrated that the absence of complement regulation in the kidney, mainly in the proximal convoluted tubule (PCT) cells makes these cells more responsive to complement-mediated injury [54, 55]. Recently, one study published a kidney autopsy report with strong C5b-9 staining and demonstrated that C5b-9 staining was prominent on the apical brush border of tubular epithelial kidney cells along with deposition on glomeruli and capillaries and nucleocapsid as well as the spiked protein of SARS-CoV-2 [56].
This hypothesis also supported that SARS-CoV infection is associated with complement regulators of two genetic variants, CD55 (known as DAF) and FH [57]. Earlier it has been allied to the atypical haemolytic uremic syndrome (aHUS), age-related macular degeneration (AMD), and haploinsufficiency of complement regulatory protein [58]. Even though these associations are required to be assessed in independent cohort studies, they must be proposed the lectin activation and alternative pathways of the complement system in COVID-19 patients. Complement inhibition has offered an effective medication in COVID-19 patients.
Auto-immune and metabolic disorders may affect the prophylaxis of COVID-19 Patients.
Some types of diseases like autoimmune & metabolic disorders, cancers, and infection have inflammatory conditions already. Elderly individuals, along with those pre-existing conditions, have shown in table 2, have demonstrated a higher possibility for developing severe COVID-19 conditions in addition to suffering a high risk of mortality and facing more chance of developing long-term complications. This pre-existing immunomodulation may affect the COVID-19 treatment by interfering with the DMPK of drugs in the COVID-19 medication. Results suggested, genetically and chemically induced model of various diseases mentioned in table 2, and analysis of transporters and CYPs in disease state is highly reasonable that dysregulation of intestinal and hepatic transporters expression and CYPs contribute to inconstant absorption and disposition of xenobiotics. That may be responsible for interpatient venerability in drug PK and therapeutic reaction [59, 60].
Table 1: Drugs that have been used for the treatment of COVID-19 patients.
| Anti-Inflammatory drugs | Dexamethasone, Ruxolitinib Baricitinib Imatinib, Tocilizumab, Itolizumab, Maplazumab, Sarilumab |
| Anti-Complement drugs | Eculizumab, AMY101 |
| Antiviral-drugs | Remdesivir, Favipiravir, Ribavirin, Interferon, Lopinavir, Ritonavir, Darunavir, Atazanavir, Umifenovir, Sofosbuvir, Daclatasvir, Molnupiravir, Teicoplanin, LCB1 |
| Antibiotics | Tetracyclines, Azithromycin |
| Others | Chloroquine, Hydroxy chloroquine, Ivermectin, ARDS-003, Fluvoxamine |
Table 2: Pre-existing immunomodulatory diseases may affect the COVID-19 treatment.
| Diseases | Immunomodulatory effect | References |
|---|---|---|
| Rheumatoid Arthritis (RA) | IL-1β, IL-6 TNF-α and IFN-γ | [61] |
| Inflammatory bowel disease (IBD) | IL-1β, IL-6 TNF-α and IFN-γ | [62, 63] |
| Chronic renal failure (CRF) | IL-1β, IL-6 TNF-α and IFN-γ | [64] |
| Diabetes (T1DM, T2DM) | IL-1β, IL-6 TNF-α, IFN-γ and CRP | [65-68] |
| Obesity | IL-1β, IL-6 TNF-α, IFN-γ and CRP | [65-68] |
| Human Immune Virus (HIV) | IL-1β, IL-6 TNF-α, IFN-γ | [59] |
| Cancer | IL-1β, IL-6, IL-23 and TNF-α, | [69] |
| Age related macular degeneration | TNF-α, IL-1 β, IL-2, IL-4, IL-6, IL-10 and Complement protein | [70] |
| atypical hemolytic uremic syndrome (aHUS) | IL-6 and Complement proteins | [71] |
| Paroxysmal Nocturnal Hemoglobinuria (PNH) | IL-6 and Complement proteins | [71] |
Alteration of drug transporters in response to immunomodulation
In the acute immune response, proinflammatory cytokines such as IL-1β, IL-6, and TNF-α play an important role [72]. These locally forming immunomodulators disseminate in the circulatory system and generate a systemic effect by binding to cell membrane receptors on the vascular endothelium and parenchymal cells of the various tissues of the human body. During infection, the immune reaction represents a significant effect in chronic diseases, which are clearly involved in modifying drug elimination (clearance) by changing the absorption and metabolism activity of therapeutic drugs. Recently, some pre-clinical model studies also reviewed the absorption, downregulation of ATP binding cassette (ABC) and solute carrier (SLC) in certain chronic inflammatory conditions [73].
P-glycoprotein (Pgp)/ABC and BCRP (breast cancer resistance protein) efflux transporters are vital proteins involved in the efflux of drugs. In addition, Pgp/ABC has been widely studied against the response of immunomodulation or inflammation. In humans, Pgp expression has been shown downregulated in intestinal epithelium cells in inflammatory conditions [74]. Human enterocyte cell line Caco-2 is used as a universal model for permeability research. It has been shown that Caco-2 was treated in conjunction with TNF-α showed downregulation of expression as well as Pgp activity [75, 76]. It is well documented about the association of Pgp mRNA level and protein expression diminished on the administration of IL-6 directly in invivo in mice or hepatic cell lines [77, 78]. Similar to Pgp, primary human hepatocytes cells also showed downregulation of BCRP expression as well as activity upon treatment with IL-6, and when these cells were treated with TNF-α this leads to the production of BCRP [79]. The treatment of IFN-γ, substantially decreased the mRNA expression of BCRP in primary human hepatocytes [80]. The human brain cell line showed a significant decrease in the BCRP expression and activity after treatment with IL-1β, IL-6, and TNF-α [81]. Proinflammatory cytokines are also thought to be important mediators of MRP2, as evidenced by the fact that IL-1β, IL-6, and TNF-α all significantly downregulate mRNA and protein expression of MRP2 in rat and human hepatic cell lines [82, 83]. The acute inflammatory response arbitrates changes in the expression of Pgp, BCRP, MRP2, and a few other important transporters [64, 76, 84-87].
The human repertoire of solute carrier (SLC) transporters as a means of tackling the immune-mediated disease. A study on inflammatory bowel diseases (IBD) patients showed that SLC transporters were altered due to inflammation [82], and mRNA expression level of SLC transporters (OATP2B1, OATP4A1, ENT1, ENT2, CNT2) in IBD patients was found dysregulated [82]. SLC expression is linked with the inflammation of the tissue and gives an indication that inflammatory signaling is involved in the regulation of disease [82]. Other than intestinal, the hepatic transporters are also involved in drug clearance. Considerable downregulation has been reported in hepatic transporters such as OATPB1, OCTN1, as well as Pgp and BCRP, after Citrobacter rodentium infection in mice as compared to control at mRNA levels [83]. This effect was not shown IL-6 or TNF-? lacking infected mice [83], again indicating the key role of these proinflammatory cytokines in controlling the expression and function of hepatic ABC and certain SLC transporters. Human hepatocytes have shown the downregulation of various transporters after facing the exposure of IL-6 [84]. These findings suggested that chronic inflammation can inhibit the expression and function of Pgp, ABC, MRP, and SLC transporters involved in drug pharmacokinetics and may occur in the intestine and in the liver as well, leading to decreased absorption [85]. Due to the inflammation, several transcription factors get activated and play a crucial role in the alteration of DMEs and transporters [86-90]. Several reviews have already available which explained the role of inflammation in the molecular mechanism of the transporters have been explained in the literature [88, 91, 92].
Alteration of drug-metabolizing enzymes in response to immunomodulation
In DMEs, CYPs are extensively engaged as the leading contributor to metabolic biotransformation of more than 60% of drugs [93]. Like drug transporters, reduced expression of CYPs and UDPGT has also been reported with the inflammatory response [73]. Immunomodulatory induced changes in hepatic CYPs expressions and activity are affected by signal molecules which are mainly cytokines, produced while progressing inflammation [94]. The regulation is an important factor in drug interactions because drug PK eventually will be altered based on disease type and produced immune response components, as well as the administered drugs [95, 96]. Many studies have described the immunomodulation (especially cytokine) induced CYPs activity modulation in different mammals [97-99] including humans [100, 101] in vitro in hepatocytes as well as in vivo in mice [102], rats [103], and humans [104]. Various diseases models like colitis [105, 106], and arthritis [107-109] have exhibited the reduction in the expression and activity of various hepatic CYPs. This inhibition exists to correlate with the rise in the concentration of proinflammatory cytokines (IL-6, TNF-α, IL-1β, IFN-γ, IL-2, and IL-1α) [105, 106, 108, 110]. Most proinflammatory cytokines are IL-1 [111-113], IL-6, TNF-α, and IFN-γ that have displayed suppression of CYPs expression and activity [114-116]. Other cytokines IL-2 and IL-10 also showed the same effect [117-119].
IL-6 is most abundantly present in the blood circulation of infected mice [105] and represses CY3A4 expression [120] and reduces CYP3A-dependent clearance of substrate drugs in the invitro culture of human hepatocyte cells [121]. To prove this hypothesis, human hepatocytes cells were treated with an IL-6 monoclonal antibody that prevented the decrease in IL-6 induced CYP3A clearance [121] and delivered the proof of IL-6 dependent CYP3A inhibition. Another study confirmed the inhibition after deletion of IL-6 encoding gene to prevent the downregulation of CYP2D and CYP3A in Clostridium rodentium infected mice [122]. One study showed that in Rat hepatocytes, human recombinant IL-6 has shown concentration-dependent inhibition of phenobarbital mediated induction of CYP2B1/2 [123] and significantly suppressed the expression of CYP1A1, CYP1A2, and CYP3A4 at mRNA levels in different human hepatoma cell lines [124]. Chronic inflammatory response, either induced by turpentine or bacterial lipopolysaccharide, in rats, showed significant inhibition in hepatic CYP1A2, CYP2A5, CYP3A11, and CYP2C1 enzymes [125, 126]. Several studies have shown that malignancies also have impacted the action of IL-6 on CYPs [127]. The role of IL-6 in cancer-mediated inhibition of hepatic CYP3A has been shown by producing such effect via monoclonal antibody against IL-6 [128] or IL-6 receptor [129] and this was also confirmed in IL-6 treated primary human hepatocytes by using IL-6 monoclonal antibody. Expression and activity of CYP1A1, CYP1A2, CYP2B6, and CYP3A4 enzymes were inhibited by IL-6, and it was also significant in intervening CYP1A2 and CYP3A4, induced by omeprazole and rifampicin, respectively. IL-6 also downregulates most phase II enzymes like UDPGT [84].
TNF-α is also released abundantly in the blood of infected mice. Like IL-6, similar experimentation has suggested that TNF-α also plays role in CYP downregulation, when treated with biological medication that deactivates soluble TNF-α [130] and deletion of TNF-α receptor gene in mice infected with Citrobacter rodentium [131], prohibited the downregulation of CYP3A11 and CYP3A25. Infliximab, an immunosuppressive drug is generally used for the treatment of rheumatoid arthritis. Pre-adjuvant arthritic rats were treated with infliximab, which resulted in a significant increase in total protein content of CYP than in untreated rats [132]. These results along with hepatic in-vitro experiments were demonstrated that TNF-α significantly regulated CYPs levels (Kinloch 2011, Nygode 2010) and provides strong evidence in CYP downregulation.
IL-1β has demonstrated the inhibition of CYP3A1 protein expression by 60 % in-vitro rat hepatocyte and reduced its activity within 6 hours [133]. Numerous studies have shown that the downregulation of CYPs due to pro-inflammatory cytokines is impeded by NO synthase inhibitors [134] and proteasome inhibitors ((Lee CM, et al. (2009)). Stimulation of the NO synthase [135] is promoted by pro-inflammatory cytokines such as IFN-γ, IL-1β, and TNF-α during inflammatory responses that precede to increase NO synthesis which inhibits CYPs expression and activity. NO prompts proteasome-based CYP3A1 inhibition [133]. These results suggest that the IL-1β inhibits the functional activity of CYP in the early stage of the inflammatory process. Authors [95] investigated the effects of IL-1β, IFN-γ, TNF-α, and TGF- β on the mRNA expression level of CYP2B6 and CYP2C9 in human hepatocytes comparing with response to control. The expression of CYP2C9 and CYP2C19 are decreased while in the presence of IL-6 and TGF- β. CYP2C8 and CYP3A4 were downregulated by all cytokine treatments.
Extrahepatic CYPs were also inhibited by pro-inflammatory mediators [142-145]. Although the cytokine-induced inhibition of CYPs is not fully demonstrated, it is anticipated and strongly recommended that a decrease in CYPs mRNA strongly insisted on a transcriptional mechanism that altered various transcriptional factors [146, 147]. Aryl hydrocarbon and nuclear factor kappa B (NF-kB) receptor are the regulatory transcription factors. In humans, rats, and mice, they are actively involved in controlling the gene expression of various CYPs in response to inflammation [146-149]. Pyrrolidine dithiocarbamate is a well-known example of an NF-kB inhibitor that has the capacity to prevent the inflammatory reduction in CYP1A2 activity [150, 151]. Inflammatory stimuli influence NF-kB, which further regulates the CYPs expressions. PXR (Pregnane X Receptor) targets a variety of genes, including CYP3A4. PXR is regulated by NF-kB factors, with NF-kB activation suppressing glucocorticoid receptors (GR) and decreasing the expression of the constitutive androsterone receptor (CAR) and its related CYP gene expression [126, 152, 153]. Highly elevated cytokines and downregulation of their targets during COVID-19 infection have been shown in table 3. It is established that alteration in pharmacokinetics of the prescribed drug can alter the activity of DMEs and the expression of drug transporters, which can contribute to interindividual heterogeneity in drug efficacy and toxicity. Generally approved drugs for COVID-19 medication, and their drug interaction targets for transporters and drug-metabolizing enzymes have been shown in Tables 4 and 5, respectively.
Table 3: The regulation of transporters and drug metabolizing enzymes by proinflammatory cytokines.
| Proinflammatory Cytokines | Species | Downregulation | References | |
|---|---|---|---|---|
| Transporters | DMEs | |||
| IL-1β | Mouse | MRP2, OATP1, OATP2, BSEP | CYP1A, CYP2B, CYP2C9, CYP2C19, CYP2D, CYP3A, UGT1A | [95, 135] |
| Human | - | CYP2C8, CYP3A4 | [95, 121, 136] | |
| IL-6 | Mouse | P-gp, MRP2, OATP1, OATP2, BSEP | CYP1A2, CYP2A5, CYP2E1, CYP3A11 | [78, 125, 135, 137] |
| Human | - | CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP3A4 | [95, 121, 138] | |
| Mouse | - | CYP1A, CYP2B, CYP2C9, CYP2C19, CYP2D, CYP3A, UGT1A | [135] | |
| Human | - | CYP2B6, CYP2C8, CYP2C9, CYP3A4 | [95] | |
| Mouse | - | CYP1A, CYP2B, CYP2C9, CYP2C19, CYP2D, CYP3A, UGT1A | [139] | |
| Human | - | CYP2C8, CYP2C9, CYP2C19, CYP3A4 | [95] | |
| Mouse | MRP2, MRP3, OATP2 | CYP1A, CYP2B, CYP2C9, CYP2C19, CYP2D, CYP3A, UGT1A/td> | [135, 137] | |
| Human | - | CYP2C8, CYP3A4 | [95, 137] | |
Table 4: Transporter mediated drug-drug interaction of therapeutic agents used to treat COVID-19.
| Drug | Involvement of Drug Transporter | Reference | |
|---|---|---|---|
| 1 | Remdesivir | $Pgp $OATPB1, IOATPB1, IOATPB3, IBSEP, IMRP4, INTCP | [140] |
| 2 | Favipiravir | IOAT1, IOAT3 | [141] |
| 3 | Ribavirin | $NT, $ENT1 | [142, 143] |
| 4 | Interferons | $OAT2 | [144] |
| 5 | Lopinavir | $Pgp, $MRP1, $MRP2, $OATP1A2, $OATP1B1 IPgp, IBCRP, IOATP1B1, IOATP1B3, IOATP2B1 | [145-147] [148-151] |
| 6 | Ritonavir | $Pgp, $MRP1, $MRP2, IPgp, IMRP1I, BCRP, IOATP1A2, IOATP2B1, IOATP1B1, IOATP1B3, IOCT1, OCT2 | [147] [152] [148, 151, 153-159] |
| 7 | Chloroquine | $OATP1A2 | [160, 161] |
| 8 | Hydroxy chloroquine | IPgp, IOATP1A2 | [160-162] |
| 9 | Dexamethasone | $Pgp, $MRP2 | [163, 164] |
| 10 | Umifenovir | Data Unavailable | |
| 11 | Teicoplanin | Data Unavailable | |
| 12 | Nitazoxanide | Data Unavailable | |
| 13 | Ivermectin | $Pgp, IBCRP, IMRP1, IMRP2, IMRP3 | [165-167] |
| 14 | Atazanavir | $Pgp, $MRP1, $MRP2, IOATP2B1 | [168, 169] [150, 151, 170, 171] |
| 15 | Azithromycin | $Pgp and $MRP2, $OATP | [172, 173] |
| 16 | Darunavir | $Pgp, $OATP1A2, OATP1B1, IPgp, IOATP2B1 | |
| 17 | Ruxolitinib | $OATP1B1, and $OCT1, $NTCP, IPgp, IBCRP | [178, 179] |
| 18 | Baricitinib | $P-gp, $BCRP, $OAT3, $MATE-K | [180, 181] |
| 19 | Imatinib | $Pgp, $OATP1B3 | [182, 183] |
| 20 | Fluvoxamine | Unclear | |
| 21 | Canabinoids | $Pgp, $BCRP, $MRPs | [184] |
| 22 | Sofosbuvir | $Pgp, $BCRP | [185] |
| 23 | Daclatasvir | $Pgp, IPgp, IBCRP, IOAT1B1, IOAT1B3 | [186] |
| 24 | Molnupiravir | Data Unavailable | |
| 25 | Itolizumab | Data Unavailable | |
| 26 | Tocilizumab | Data Unavailable | |
| 27 | Meplazumab | Data Unavailable | |
| 28 | Sarilumab | Data Unavailable | |
| 28 | Eculizumab | Data Unavailable | |
| 28 | AMY101 | Data Unavailable | |
| 28 | ARDS-003 | Data Unavailable | |
| 28 | LCB1 | Data Unavailable | |
| $-substrate of, I-Inhibitor of, Pgp; P-glycoprotein, OAT; Organic Anion Transporter, OATP; Organic Anion Transporter Protein, BCRP; Breast Cancer Resistance Protein, MRP, Multidrug Resistance Associated Protein, OCT; Organic Cation Transporter, NT; Nucleotide Transporter, ENT; Equilibrative Nucleoside Transporter, NTCP; Sodium/Taurocholate Co-transporting polypeptide, BSEP; Bile Salt Export Pump, | |||
Table 5: CYP mediated drug-drug interaction of therapeutic agents used to treat COVID-19.
| Drug | Metabolism | Reference | |
|---|---|---|---|
| 1 | Remdesivir | $CYP2C8, $CYP2D6, $CYP3A4, ICYP3A4 | [140] |
| 2 | Favipiravir | $AO, ICYP2C8 | [141], [187] |
| 3 | Ribavirin | Phosphorylation, Deribosylation, Amide hydrolysis | [181] |
| 4 | Interferons | $CYP1A2, $UGT2B7, ICYP3A, ICYP2D6 | [144, 188] |
| 5 | Lopinavir | ICYP3A4 | [145-147] |
| 6 | Ritonavir | $CYP1A2, $CYP2C8, $CYP2C9, $CYP2C19, ICYP3A4, ICYP2D6 | [189] [152] |
| 7 | Chloroquine | $CYP2C8, $CYP3A4 $CYP2D6 | [190, 191] |
| 8 | Hydroxy chloroquine | $CYP2C8, $CYP3A4 $CYP2D6 | [190-192] |
| 9 | Dexamethasone | $CYP3A4 | [193] |
| 10 | Umifenovir | $CYP3A4 and $FMOs | [194] |
| 11 | Teicoplanin | Unclear Metabolic path | [195] |
| 12 | Nitazoxanide | $Deacetylase, $UGT | [196] |
| 13 | Ivermectin | ICYP2C9, ICYP2C19 ICYP2D6, ICYP3A4 | [197] |
| 14 | Atazanavir | ICYP3A4, IUDGT | [168] |
| 15 | Azithromycin | $CYP3A4 | [198, 199] |
| 16 | Darunavir | ICYP3A4 | [178, 179] |
| 17 | Ruxolitinib | $CYP1A2, $CYP2B6, $CYP2C9 $CYP3A4 | [200, 201] |
| 18 | Baricitinib | $CYP3A4 | [181, 202] |
| 19 | Imatinib | $CYP2C8, $CYP3A4, ICYP3A4 | [203, 204] |
| 20 | Fluvoxamine | $CYP1A2, $CYP2C19, $CYP2D6, $CYP3A4 ICYP1A2, ICYP2C19 | [205, 206, 207] |
| 21 | Canabinoids | $CYPs, $UGT | [184, 208] |
| 22 | Sofosbuvir | $Hydrolases | [209] |
| 23 | Daclatasvir | $CYP3A4 | [186] |
| $-Substrate of, I-Inhibitor of, CYP; Cytochrome P450, UGT, UDP-Glucuronosyltransferase, AO; Aldehyde Oxidase, FMO; Flavin-containing Monooxygenase | |||
Consequences of DDI of a used drug in COVID-19 treatment
CYP isoforms, particularly CYP1A2, CYPC9, CYP2C19, CYP2D6, and CYP3A4, are responsible for approximately 80% of all drug metabolism [228]. Several CYP450 isoenzymes, including CYP1A2, CYP2B6, CYPC9, CYP2C19, CYP2D6, and CYP3A4, have had their mRNA expression reduced by the inflammatory response suggested by in-vitro data [45]. As a result, theoretically, inflammation may have an impact on their pharmacokinetics. For medications with a restricted therapeutic index, such as hydroxychloroquine and chloroquine, this point is critical for clinical relevance.
Inflammation effect on the PK of antiretrovirals, which are primarily metabolized by CYP3A4, would be expected. For COVID-19 treatment lopinavir, ritonavir, atazanavir, and darunavir drugs have been used. These drugs are either substrate or inhibitor of different transporters (P-gp, OATP1B1, and BCRP) and their metabolism is mediated by CYP3A4. In HIV-positive patients, CYP3A activity was approximately 50% lower than in healthy volunteers [229]. In HIV patients, there was no significant association between inflammatory biomarker concentration and atazanavir clearance [230]. Later in this study authors also showed that the presence of booster ritonavir, which is consistently connected with atazanavir to limit its clearance, may have reduced the effect of inflammation. Two recent short reports found increased plasma lopinavir concentrations in severe COVID-19 patients [231, 232], relative to those found in HIV patients, and in relation to inflammation [231]. Additional evidence supporting changes in PK of medication based on HIV-serostatus includes lower concentrations of atazanavir in HIV patients compared to healthy subjects and higher concentrations of darunavir in HIV patients compared to healthy volunteers [233].
Remdesivir has a linear PK profile. In COVID-19 patients, an intravenous administration, exhibited a peak at the end of infusion, while its phosphorylated metabolite GS-441524 reached a peak and then remained detectable with a half-life of more than 35 hours until the next remdesivir administration [210]. While there was no significant variation in the half-life of remdesivir in healthy adults, the nucleoside metabolite GS-441524 had a half-life of 24 hours [211]. Remdesivir is significantly metabolized by CYP2C8, CYP2D6, and CYP3A4 according to primary data from healthy human donors [158]. Remdesivir is a substrate of P-gp, active efflux in the lungs and CNS could potentially be involved. Remdesivir was found below the detection range in all the compartments tested, although GS-441524 was found in bronchoalveolar aspirate and a low concentration was also detected in CSF [210]. When chloroquine or hydroxychloroquine is used with remdesivir, the antiviral activity of remdesivir may be reduced [236]. Co-treatment of remdesivir with baricitinib, a CYP3A4 substrate, resulted in a significant adverse effect despite a rapid improvement in clinical status [237]. Although no scientific studies on remdesivir's DDI have been completed, a mathematical predication of DDI liability has been made utilizing current phase I and in-vitro data [238]. Valuation for the remdesivir potential to prevent transporters and DMEs in-vivo, a physiologically based pharmacokinetic (PBPK) model is developed to capture the in vitro inhibition potency of remdesivir and in vivo PK using SimCYP software. Because of the high to moderate extraction ratio (0.6 to 0.8) and IV route of administration, when remdesivir had a low hepatic extraction ratio the effect of inducer/inhibitor on the PK of remdesivir will be significantly reduced [239]. The variance in midazolam (CYP3A), rosuvastatin (OATP/BCRP), metformin (MATE1), and pravastatin (OATP) probe exposure (AUC) was predicted using in vitro unbound inhibition constants for CYP3A, BCRP, OATP1B3, and MATE1. For these PBPK simulations, the time of remdesivir delivery relative to prob drug was optimized to predict the maximum DDI, which was roughly related to the administration of remdesivir, so the infusion ended at the probe maximum observed plasma concentration. At therapeutic remdesivir doses, co-administration of remdesivir is thought to enhance probing drug AUC by transporters. Previous studies showed that a 200 mg loading dose followed by 100 mg maintenance doses for roughly 5 to 10 days resulted in a stable PK for the COVID-19 medication [212].
Favipiravir is an antiviral prodrug that is converted to an active metabolite through phosphorylation by the intracellular enzyme hypoxanthine-guanine phosphoribosyl transferase (HGPRT). It shows non-CYPs facilitated biotransformation by aldehyde oxidase (AO) and xanthine oxidase (XO). Variants of AO are frequently linked to pharmacodynamics (PD) in other medications that are AO's substrate. Because of the genetic variability of AO, elevated plasma levels of favipiravir must be indicated in Asian populations. Based on AO, the DDI of zaleplon and cimetidine is already known. Cimetidine co-administration inhibits AO-catalyzed oxo-zaleplon formation, and the Zaleplon level is cautiously included. Clinicians should be aware that co-administration of the medicine with an AO inhibitor, such as cimetidine, tamoxifen phenothiazines, verapamil, amlodipine, nifedipine, loratadine, cyclobenzaprine, ondansetron, or ketoconazole, could theoretically increase plasma levels of favipiravir active metabolites because of possible drug-drug interactions. Its metabolite inhibits OAT1 and OAT3 in a moderate way [141]. It is also a moderate inhibitor of various CYPs, but its effect on CYP2C8 must be clinically significant to enhance exposure [187] to drugs like rosiglitazone (hypoglycemic), repaglinide, torasemide, paclitaxel, and buprenorphine.
The hydroxychloroquine (HCQ) metabolism is hepatic but not been precisely characterized and the effect CYP2C8 CYP2D6 and CYP3A4/5 being extrapolated from chloroquine (CQ) data. The half-life of HCQ elimination is approximate 40 days. Both CQ and HCQ transform into active metabolites by CYP isomers through the dealkylation process [213, 214].
Systemic Lupus Erythematosus (SLE) patients demonstrated that HCQ and CQ both have variability in metabolism and the effect of CYP2D6 SNPs on blood HCQ level [215]. The gene polymorphisms of CYP2C8 CYP2D6 and CYP3A4/5 may also alter the disposition. As immunologically challenged patients frequently developed more severe COVID-19 clinical complications. HCQ medication treatment for COVID-19 infection was contested by various side effects among individual variability in CYP genotypes. Specifically, CYP2D6 genotyping may be helpful to decide the best possible HCQ dosage in the context of personalized medicine. CQ and HCQ are both inhibitors of P-gp. Both drugs also increase cyclosporine levels, with HCQ increasing digoxin levels. The multiple DDI that are now known, as well as the potential use of these agents in combination with other drug therapies, requires consideration for the safety of the patients.
Azithromycin is an antibiotic belonging to the macrolide group, used to prescribe COVID-19 patients who developed the risk of secondary infection. It has high tissue affinity with wide distribution in the body. The elimination half-life of azithromycin is between 2-4 days. It is primarily eliminated via the liver. It is noticed mainly in the bile and urine as a parental form. It inhibits CYP3A4 [198], OATP1A2 and OATP2B1 [173] moderately. Azithromycin and HCQ both drugs are both metabolized by CYP3A4. This combination was revealed to decrease the mortality associated with COVID-19 infection [216]. Co-medication of azithromycin heightened the QT of HCQ results enhance the chances of cardiac failure and cardiovascular mortality [217]. The possibility of a probable interaction is not PK but PD while recommending azithromycin as co-medication in connection with its effect on QT prolongation. Risks should be evaluated when azithromycin is administered to COVID patients with impaired hepatic function.
Dexamethasone is a synthetic glucocorticoid. It is 6-hydroxylated (6α- and 6β-hydroxy dexamethasone) by CYP3A4 and reversibly metabolized to dexamethasone by 11β- dehydrogenase isozyme. Dexamethasone is an agonist of nuclear pregnane X receptor (PXR) [218] and It is a weak inducer of CYP3A4 [219, 220] through PXR, with most data indicating that it decreases the exposure of sensitive CYP3A4 substrates by approximately 20%. PXR also changed the expression of other drug-metabolizing enzymes and transporters, such as CYP3A11, CYP2B10, and OATP2 [221-223]. Drug metabolizing enzyme variants (CYP3A4, CYP3A5, CYP3A7, and GSTT1) and transporters (ABCB1 and MDR1) have been associated with response to corticosteroids in several diseases [224]. Overdoses of corticosteroids are considered sufficiently immunosuppressive to warrant unease about probably lessening the effectiveness of vaccines. Corticosteroids also have a high risk of producing or aggravating hyperglycemia and may diminish the efficacy of antidiabetic drugs.
Ivermectin is presented as necessary medicine of the WHO model list. Ivermectin is mostly metabolized by CYP3A4, and it inhibits CYPC9, CYP2C19, CYP2D6, and CYP3A4 [197]. In animals, Ivermectin DDIs begin mainly at the level of P-gp (ABCB1). It is a substrate for P-gp [225] that enables ivermectin intestinal and biliary excretion and prevents it from entering the CNS (Central Nervous System) [226, 227]. Other transporters might also be involved in ivermectin DDIs like MRPs and BCRP [228]. Pgp and CYP3A4 inhibitors may increase the ivermectin level in the plasma. Ideally, metabolism might decrease with age leading to higher exposure of ivermectin in aged patients [229].
Umifenovir is also described in the publication as Arbidol. It was mainly excreted in urine as phase II conjugates with glucuronide and glucuronide sulfinyl as major metabolites. Arbidol is a substrate for CYP3A4 and FMOs in vitro, insinuating that use with CYP3A4 inducer and inhibitors may lead to possibly significant increases and decreases in arbidol exposure. Total 33 arbidol metabolites were found in human plasma, urine, and feces by engaging the different CYPs including CYP1A1, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, FMO1, FMO3, and FMO5 [194]. Three major metabolites (M5, M6-1, and M8) were detected in the plasma following oral treatment of umifenovir to healthy individuals. M6-1 is critical to assess for safety and efficacy because of its high exposure and extended elimination half-life. CYP3A4 was the most active enzyme in the arbidol metabolism in the liver and gut, followed by other CYPs and FMOs [194]. However, concern and close monitoring are advisable when it is on medication, and medical practitioners are reassured to inform presumed DDI that they observe concerning arbidol.
Ruxolitinib and baricitinib, the Janus kinase inhibitor that has been used to treat COVID-19. Ruxolitinib is mostly metabolized by CYP3A4 and CYP2C9, with CYP1A2 and CYP2B6 playing a minor role [200, 201]. Following inhibition of both CYP2C9 and CYP3A4 enzymes, fluconazole may increase plasma exposure. On the basis of the PBPK model, the importance of this interaction was validated in healthy subjects [230]. Fluconazole simulation findings were used as a foundation for ruxolitinib dose correction when perpetrator drugs were co-administered. Baricitinib clinical drug-drug interaction still needs to be known. It is metabolized in part by CYP3A4 and is a substrate of P-gp, BCRP, OAT3, multidrug, and toxin extrusion protein (MATE) 2K. OCT1 is likewise inhibited by baricitinib. Probenecid is used for treating gout disease, increasing the exposure of baricitinib following inhibition of OAT3, and lowering the renal clearance in healthy participants. The renal clearance and inhibitory effect of probenecid on baricitinib were reproduced in vitro using PBPK modelling [180]. Both have immunosuppressive effects and associated interactions concerns regarding other immunosuppressants and live vaccines.
Imatinib is the tyrosine kinase inhibitor and is solely metabolized by CYP3A4 and CYP2C8 [204]. It was also shown that CYP3A4 inhibition occurs through an irreversible mechanism [231]. The time-dependent auto inhibition of its own CYP3A4 metabolism leads to an important role for CYP2C8 in the elimination of imatinib. The auto inhibition of its own CYP3A4 metabolism in a time-dependent manner resulted in a significant involvement for CYP2C8 in imatinib elimination. Interactions with other drugs and pharmacogenetic polymorphisms both impact CYP2C8 activity during many administrations, resulting in apparent interindividual variability in imatinib exposure. Dexamethasone is a potent inducer of CYP3A4 and significantly reduces the plasma level of imatinib. However, there is no literature available to find DDI with imatinib and dexamethasone. Imatinib is a known substrate of ABC transporters [232, 233], they have been reported to antagonize these transporters. This effect could manipulate the PK properties of imatinib, perhaps lessening its absorption subsequently causing imatinib concentration to drop below therapeutic levels [234, 235].
Fluvoxamine is a serotonin reuptake inhibitor with a half-life of approximately 30 hours. It has anti-inflammatory properties. It is known that it inhibits the enzyme CYP1A2. It is, however, a strong inhibitor of CYP2C19. A case study of a 48-year-old lady with a psychiatric disorder has died following treatment of multiple drugs along with fluvoxamine. After the autopsy, femoral and cardiac blood, urine, and bile were collected for toxicological kinetics. Fluvoxamine, propranolol, clotipine, gabapentin, 7-aminoclonazepam, and haloperidol drugs were detected after the examination. Fluvoxamine levels in the blood exceeded the upper limit of therapeutic blood level by approximately ten times. The relevant cause of death was due to multiple drug use [236]. Fluvoxamine has a minor effect on medications that are metabolized by CYP3A4. It is classified as a potent inhibitor of CYP1A2 and CYP2C19 by the FDA. It is commonly used for clinical DDI [237]. PBPK model was created to characterize the CYP1A2 and CYP2C19 for interaction between fluvoxamine and other drugs [238]. As a result, it can be used to support prospective dose adaptation suggestions, labelling, and clinical DDI trial design.
Sofosbuvir is a substrate of Pgp and BCRP [185]. Hydrolases metabolize sofosbuvir extensively, and GS-331007 metabolite accounts for more than 90 % of overall drug exposure [209]. The metabolizing enzymes and drug transporters are not inhibited or induced by sofosbuvir or its metabolites. Daclatasvir is a substrate for CYP3A4 Pgp and also the inhibitor of Pgp, BCRP, OATP1B1, and OATP1B3 [186]. The feces are the predominant route of elimination for daclatasvir, followed by the kidneys. Sofosbuvir and daclatasvir have exhibited good safety profiles with minimum drug interaction and widely available therapeutic options for COVID-19 treatment.
Some of the drugs included in Table 1 lack pharmacogenomic data and are still in clinical trials for the treatment of COVID-19 infection, therefore more research is needed to determine drug metabolism and disposition in the inflammatory and immunomodulatory state.
Conclusion
COVID-19 is classified as a multisystemic disease. The elementary pathogenesis involves distinct components; immune deficiency and severe lung inflammation, which leads to increased production of cytokines and is related to inappropriate immune response. Timely management of the cytokine storm in its early stage through immunomodulators, cytokine antagonists as well as the reduction of lung inflammatory cell infiltration, are the key to reducing the mortality rate and improving the treatment success rate of COVID-19 patients. In consequence, treatment approaches at present include anti-infectious, anti-viral, anti-proinflammatory cytokines, and life support therapies. The potential of disease-drug or drug-drug interaction is an essential concern while providing optimal treatment regimens for individual patients. Hence, the present review expounded the probability of drug interaction in COVID-19 patients while reaching inflammatory conditions and or having comorbidity like autoimmune and metabolic diseases. This point is highly clinical relevance for drugs with a narrow therapeutic index like HCQ and CQ. As the mechanism underlying the observed alteration in the plasma binding proteins, drug transporters, and DMEs comes to be clear, the next step is to address the clinical importance to better predict the drug concentration in the inflammatory state of the disease condition. Proinflammatory cytokines impede the expression and activity of drug transporters and DMEs (hepatic and extrahepatic). Alteration in the transporters and DMEs can lead to changes in the PK parameters of used drugs. The risk of drug interactions should not be prohibited since they are frequently manageable and convenient. The consumption of a single drug may possibly not be more effective, but during co-medication of multiple drugs, the risk of drug interaction must be increased. The impact of inflammation on the PK of the drug remains and alternated an important field of research.
Funding
Authors have not received any funding from any private and government organization.
Conflict of Interest
The author have no conflict of interest.
Overlapping Publication
This article have not been submitted to any other journal for publication.
References
1. Helmy YA, Fawzy M, Elaswad A, et al. (2020) The COVID-19 pandemic: A comprehensive review of taxonomy, genetics, epidemiology, diagnosis, treatment, and control. J Clin Med 9: 1225.
https://doi.org/10.3390/jcm9041225
2. Huang C, Wang Y, Li X, et al. (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395: 497-506. https://doi.org/10.1016/S0140-6736(20)30183-5
3. Don BR, Kaysen G (2004) Serum albumin: relationship to inflammation and nutrition. Semin Dial 17: 432-437. https://doi.org/10.1111/j.0894-0959.2004.17603.x
4. HJ Moshage, JA Janssen, JH Franssen, et al. (1987) Study of the molecular mechanism of decreased liver synthesis of albumin in inflammation. J Clin Invest 79: 1635-1641.
https://doi.org/10.1172/JCI113000
5. B Ruot, F Béchereau, G Bayle, et al. (200) The response of liver albumin synthesis to infection in rats varies with the phase of the inflammatory process. Clin Sci (Lond) 102: 107-114.
https://doi.org/10.1042/cs1020107
6. RR Shah, RL Smith (2015) Inflammation-induced phenoconversion of polymorphic drug metabolizing enzymes: hypothesis with implications for personalized medicine. Drug Metab Dispos 43: 400-410.
https://doi.org/10.1124/dmd.114.061093
7. Morgan ET (2009) Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther 85: 434-438.
https://doi.org/10.1038/clpt.2008.302
8. ET Morgan, KB Goralski, M Piquette-Miller, et al. (2008) Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab Dispos 36: 205-216. https://doi.org/10.1124/dmd.107.018747
9. G Grasselli, A Pesenti, M Cecconi (2020) Critical care utilization for the COVID-19 outbreak in lombardy, italy: early experience and forecast during an emergency response. JAMA 28: 1545-1546. https://doi.org/10.1001/jama.2020.4031
10. HJ Mann (2006) Drug-associated disease: cytochrome P450 interactions. Crit Care Clin 22: 329-345.
https://doi.org/10.1016/j.ccc.2006.02.004
11. Kumar D, Trivedi N (2021) Disease-drug anddrug-drug interaction in COVID-19: risk and assessment. Biomed Pharmacother 139: 111642. https://doi.org/10.1016/j.biopha.2021.111642
12. N Trivedi, A Verma, D Kumar (2020) Possible treatment and strategies for COVID-19: review and assessment. Eur Rev Med Pharmacol Sci 24: 12593-12608.
https://doi.org/10.26355/eurrev_202012_24057
13. Mitja O, Clotet B (2020) Use of antiviral drugs to reduce COVID-19 transmission. Lancet Glob Health 8: e639-e640. https://doi.org/10.1016/S2214-109X(20)30114-5
14. Rismanbaf A (2020) Potential treatments for COVID-19; a narrative literature review. Arch Acad Emerg Med 8: e29.
15. Ledford H (2020) Coronavirus breakthrough: dexamethasone is first drug shown to save lives. Nature 582: 469. https://doi.org/10.1038/d41586-020-01824-5
16. Kasgari HA, Moradi S, Shabani AM, et al. (2020) Evaluation of the efficacy of sofosbuvir plus daclatasvir in combination with ribavirin for hospitalized COVID-19 patients with moderate disease compared with standard care: a single-centre, randomized controlled trial. J Antimicrob Chemother 75: 3373-3378. https://doi.org/10.1093/jac/dkaa332
17. EJ Lenze, C Mattar, CF Zorumski, et al. (2020) Fluvoxamine vs Placebo and clinical deterioration in outpatients with symptomatic COVID-19: A randomized clinical trial. JAMA 324: 2292-2300. https://doi.org/10.1001/jama.2020.22760
18. RM Cox, JD Wolf, RK Plemper (2020) Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nat Microbiol 6: 11-18. https://doi.org/10.1038/s41564-020-00835-2
19. SA Grupp, M Kalos, D Barrett, et al. (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368: 1509-1518. https://doi.org/10.1056/NEJMoa1215134
20. AK Azkur, M Akdis, D Azkur, et al. (2020) Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 75: 1564-1581. https://doi.org/10.1111/all.14364
21. J Hadjadj, N Yatim, L Barnabei, et al. (2020) Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369: 718-724 https://doi.org/10.1126/science.abc6027
22. X Li, M Geng, Y Peng, et al. (2020) Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal 10: 102-108. https://doi.org/10.1016/j.jpha.2020.03.001
23. Lucas C, Wong P, Klein J, et al. (2020) Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584: 463-469. https://doi.org/10.1038/s41586-020-2588-y
24. McElvaney OJ, McEvoy NL, McElvaney OF, et al. (2020) Characterization of the inflammatory response to severe COVID-19 illness. Am J Respir Crit Care Med 202: 812-821. https://doi.org/10.1164/rccm.202005-1583oc
25. Nile SH, Nile A, Qiu J, et al. (2020) COVID-19: Pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev 53: 66-70. https://doi.org/10.1016/j.cytogfr.2020.05.002
26. Wilson MP, Jack AS (2020) Coronavirus disease 2019 (COVID-19) in neurology and neurosurgery: A scoping review of the early literature. Clin Neurol Neurosurg 193: 105866. https://doi.org/10.1016/j.clineuro.2020.105866
27. Ronit A, Berg RMG, Bay JT, et al. (2021) Compartmental immunophenotyping in COVID-19 ARDS: A case series. J Allergy Clin Immunol 147: 81-91. https://doi.org/10.1016/j.jaci.2020.09.009
28. Fajgenbaum DC, June CH (2020) Cytokine storm. N Engl J Med 383: 2255-2273. https://doi.org/10.1056/nejmra2026131
29. Moore JB, June CH (2020) Cytokine release syndrome in severe COVID-19. Science 368: 473-474.
30. Esteban YM, de Jong JLO, Tesher MS (2017) An overview of hemophagocytic lymphohistiocytosis. Pediatr Ann 46: e309-e313. https://doi.org/10.3928/19382359-20170717-01
31. Vabret N, Samstein R, Fernandez N, et al. (2020) Advancing scientific knowledge in times of pandemics. Nat Rev Immunol 20: 338. https://doi.org/10.1038/s41577-020-0319-0
32. Merad M, Martin JC (2020) Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol 20: 355-362. https://doi.org/10.1038/s41577-020-0331-4
33. Zhang X, Tan Y, Ling Y, et al. (2020) Viral and host factors related to the clinical outcome of COVID-19. Nature 583: 437-440. https://doi.org/10.1038/s41586-020-2355-0
34. Wu C, Chen X, Cai Y, et al. (2020) Risk factors associated with acute respiratory distress syndrome and death in patients with Coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med 180: 934-943. https://doi.org/10.1001/jamainternmed.2020.0994
35. Mehta P, McAuley DF, Brown M, et al. (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395: 1033-1034. https://doi.org/10.1016/s0140-6736(20)30628-0
36. Del Valle DM, Kim-Schulze S, Huang H, et al. (2020) An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med 26: 1636-1643. https://doi.org/10.1038/s41591-020-1051-9
37. Schulert GS, Grom AA (2015) Pathogenesis of macrophage activation syndrome and potential for cytokine- directed therapies. Annu Rev Med 66: 145-59. https://doi.org/10.1146/annurev-med-061813-012806
38. Zabetakis I, Lordan R, Norton C, et al. (2020) COVID-19: the inflammation link and the role of nutrition in potential mitigation. Nutrients 12:1466. https://doi.org/10.3390/nu12051466
39. Tay MZ, Poh CM, Rénia L, et al. (2020) The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol 20: 363-374. https://doi.org/10.1038/s41577-020-0311-8
40. Ye Q, Wang B, J Mao (2020) The pathogenesis and treatment of the `Cytokine Storm' in COVID-19. J Infect 80: 607-613. https://doi.org/10.1016/j.jinf.2020.03.037
41. Zhou F, Yu T, Du R, et al. (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395: 1054-1062. https://doi.org/10.1016/s0140-6736(20)30566-3
42. Zhang C, WU Z, Li J, et al. (2020) Cytokine release syndrome in severe COVID-19: interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int J Antimicrob Agents 55: 105954. https://doi.org/10.1016/j.ijantimicag.2020.105954
43. Xu, X, Han M, Li T, et al. (2020) Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci U S A 117: 10970-10975. https://doi.org/10.1073/pnas.2005615117
44. Sanders JM, Monogue ML, Jodlowski TZ, et al. (2020) Pharmacologic treatments for coronavirus disease 2019 (COVID-19): A review. JAMA 323: 1824-1836. https://doi.org/10.1001/jama.2020.6019
45. Stoermer KA, Morrison TE (2011) Complement and viral pathogenesis. Virology 411: 362-73. https://doi.org/10.1016/j.virol.2010.12.045
46. Liszewski MK, Bertram P, Leung MK, et al. (2008) Smallpox inhibitor of complement enzymes (SPICE): regulation of complement activation on cells and mechanism of its cellular attachment. J Immunol 181: 4199-207. https://doi.org/10.4049/jimmunol.181.6.4199
47. Tam JCH, Bidgood SR, McEwan WA, et al. (2014) Intracellular sensing of complement C3 activates cell autonomous immunity. Science 345: 1256070. https://dx.doi.org/10.1126/science.1256070
48. Gralinski LE, Sheahan TP, Morrison TE, et al. (2018) Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. MBio 9: e01753-18. https://doi.org/10.1128/mbio.01753-18
49. Java A, Apicelli AJ, Liszewski MK, et al. (2020) The complement system in COVID-19: friend and foe? JCI Insight 5: e140711. https://doi.org/10.1172/jci.insight.140711
50. Gao T, Hu M, Zhang X, et al, (2020) Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. MedRxiv PPR: PPR130597 https://doi.org/10.1101/2020.03.29.20041962
51. Shen B, Yi X, Sun Y, et al. (2020) Proteomic and metabolomic characterization of COVID-19 Patient sera. Cell 182: 59-72.E15. https://doi.org/10.1016/j.cell.2020.05.032
52. Cugno M, Meroni PL, Gualtierotti R, et al. (2020) Complement activation in patients with COVID-19: A novel therapeutic target. J Allergy Clin Immunol 146: 215-217. https://doi.org/10.1016/j.jaci.2020.05.006
53. Magro C, Mulvey JJ, Berlin D, et al. (2020) Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl Res 220: 1-13. https://doi.org/10.1016/j.trsl.2020.04.007
54. Ichida S, Y Yuzawa, H Okada et al. (1994) Localization of the complement regulatory proteins in the normal human kidney. Kidney Int 46: 89-96. https://doi.org/10.1038/ki.1994.247
55. Tang S, Sheerin NS, Zhou W, et al. (1999) Apical proteins stimulate complement synthesis by cultured human proximal tubular epithelial cells. J Am Soc Nephrol 10: 69-76. https://doi.org/10.1681/asn.v10169
56. Diao B, Wang C, Wang R, et al. (2021) Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat Commun 12: 2506. https://doi.org/10.1038/s41467-021-22781-1
57. Ramlall V, Thangaraj PM, Meydan C, et al. (2020) Identification of immune complement function as a determinant of adverse SARS-CoV-2 infection outcome. MedRxiv 5.20092452. https://doi.org/10.1101/2020.05.05.20092452
58. Liszewski MK, Java A, Schramm EC, et al. (2017) Complement dysregulation and disease: Insights from contemporary genetics. Annu Rev Pathol 12: 25-52. https://doi.org/10.1146/annurev-pathol-012615-044145
59. Cressman AM, Petrovic V, Piquette-Miller M (2012) Inflammation-mediated changes in drug transporter expression/activity: implications for therapeutic drug response. Expert Rev Clin Pharmacol 5: 69-89. https://doi.org/10.1586/ecp.11.66
60. Stanke-Labesque F, Gautier-Veyret E, Chhun S, et al. (2020) Inflammation is a major regulator of drug metabolizing enzymes and transporters: Consequences for the personalization of drug treatment. Pharmacol Ther 215: 107627. https://doi.org/10.1016/j.pharmthera.2020.107627
61. Choy EH, Panayi GS (2001) Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med 344: 907-16. https://doi.org/10.1056/nejm200103223441207
62. Yan Y, Kolachala V, Dalmasso G, et al. (2009) Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS One 4: e6073. https://doi.org/10.1371/journal.pone.0006073
63. Raddatz D, Bockemuhl M, Ramadori G (2005) Quantitative measurement of cytokine mRNA in inflammatory bowel disease: relation to clinical and endoscopic activity and outcome. Eur J Gastroenterol Hepatol 17: 547-57. https://doi.org/10.1097/00042737-200505000-00012
64. Mak RH, Cheung W, Cone RD, et al. (2006) Mechanisms of disease: Cytokine and adipokine signaling in uremic cachexia. Nat Clin Pract Nephrol 2: 527-34. https://doi.org/10.1038/ncpneph0273
65. Pickup JC, Mattock MB, Chusney GD, et al. (1997) NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 40: 1286-92. https://doi.org/10.1007/s001250050822
66. Pradhan AD, Manson JE, Rifai N, et al. (2001) C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 286: 327-34. https://doi.org/10.1001/jama.286.3.327
67. Esposito K, Nappo F, Marfella R, et al. (2002) Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation 106: 2067-72. https://doi.org/10.1161/01.cir.0000034509.14906.ae
68. Spranger J, Kroke A, Möhlig M, et al. (2003) Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-potsdam study. Diabetes 5: 812-7. https://doi.org/10.2337/diabetes.52.3.812
69. Mantovani A, Allavena P, Sica A, et al. (2008) Cancer-related inflammation. Nature 45: 436-44. https://doi.org/10.1038/nature07205
70. Litwinska Z, Sobu? A, ?uczkowska K, et al. (2019) The interplay between systemic inflammatory factors and MicroRNAs in age-related macular degeneration. Front Aging Neurosci 11: 286. https://doi.org/10.3389/fnagi.2019.00286
71. Chauhan AJ, Wiffen LJ, Brown TP (2020) COVID-19: A collision of complement, coagulation and inflammatory pathways. J Thromb Haemost 18: 2110-2117. https://doi.org/10.1111/jth.14981
72. Furuta Y, Gowen BB, Takahashi K, et al. (2013) Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res 100: 446-54. https://doi.org/10.1016/j.antiviral.2013.09.015
73. Wu KC, Lin CJ (2019) The regulation of drug-metabolizing enzymes and membrane transporters by inflammation: Evidences in inflammatory diseases and age-related disorders. J Food Drug Anal 27: 48-59. https://doi.org/10.1016/j.jfda.2018.11.005
74. Blokzijl H, Borght SV, Bok LIH, et al. (2007) Decreased P-glycoprotein (P-gp/MDR1) expression in inflamed human intestinal epithelium is independent of PXR protein levels. Inflamm Bowel Dis 13: 710-20. https://doi.org/10.1002/ibd.20088
75. Belliard AM, Lacour B, Farinotti R, et al. (2004) Effect of tumor necrosis factor-alpha and interferon-gamma on intestinal P-glycoprotein expression, activity, and localization in Caco-2 cells. J Pharm Sci 93: 1524-36. https://doi.org/10.1002/jps.20072
76. Buyse M, Radeva G, Bado A, et al. (2005) Intestinal inflammation induces adaptation of P-glycoprotein expression and activity. Biochem Pharmacol 6: 1745-54. https://doi.org/10.1016/j.bcp.2005.03.025
77. Lee G, M Piquette-Miller (2001) Influence of IL-6 on MDR and MRP-mediated multidrug resistance in human hepatoma cells. Can J Physiol Pharmacol 79: 876-84.
78. Sukhai M, Yong A, Kalitsky J, et al. (2000) Inflammation and interleukin-6 mediate reductions in the hepatic expression and transcription of the mdr1a and mdr1b Genes. Mol Cell Biol Res Commun 4: 248-56. https://doi.org/10.1006/mcbr.2001.0288
79. Le Vee M, et al. (2009) Regulation of drug transporter expression in human hepatocytes exposed to the proinflammatory cytokines tumor necrosis factor-alpha or interleukin-6. Drug Metab Dispos 37: 685-93. https://doi.org/10.1124/dmd.108.023630
80. Le Vee M, Lecureur V, Stieger B, et al. (2011) Regulation of drug transporter mRNA expression by interferon-gamma in primary human hepatocytes. Fundam Clin Pharmacol 25: 99-103. https://doi.org/10.1124/dmd.108.023630
81. Poller B, Drewe J, Krähenbühl S, et al. (2010) Regulation of BCRP (ABCG2) and P-glycoprotein (ABCB1) by cytokines in a model of the human blood-brain barrier. Cell Mol Neurobiol 30:63-70. https://doi.org/10.1007/s10571-009-9431-1
82. Wojtal KA, Eloranta JJ, Hruz P, et al. (2009) Changes in mRNA expression levels of solute carrier transporters in inflammatory bowel disease patients. Drug Metab Dispos 37: 1871-7. https://doi.org/10.1124/dmd.109.027367
83. Merrell MD, Nyagode BA, Clarke JD, et al. (2014) Selective and cytokine-dependent regulation of hepatic transporters and bile acid homeostasis during infectious colitis in mice. Drug Metab Dispos 42: 596-602. https://doi.org/10.1124/dmd.113.055525
84. Keller R, Klein M, Thomas M, et al. (2016) Coordinating role of RXRalpha in downregulating hepatic detoxification during inflammation revealed by fuzzy-logic modeling. PLoS Comput Biol 12: e1004431. https://doi.org/10.1371/journal.pcbi.1004431
85. Simon F, Garcia J, Guyot L, et al. (2019) Impact of interleukin-6 on drug-metabolizing enzymes and transporters in intestinal cells. AAPS J 22: 16. https://doi.org/10.1208/s12248-019-0395-x
86. Ho EA, Piquette-Miller M (2007) KLF6 and HSF4 transcriptionally regulate multidrug resistance transporters during inflammation. Biochem Biophys Res Commun 353: 679-85. https://doi.org/10.1016/j.bbrc.2006.12.090
87. Kameyama N, Arisawa S, Ueyama J, et al. (2008) Increase in P-glycoprotein accompanied by activation of protein kinase Calpha and NF-kappaB p65 in the livers of rats with streptozotocin-induced diabetes. Biochim Biophys Acta 1782: 355-60. https://doi.org/10.1016/j.bbadis.2008.02.005
88. Teng S, Piquette-Miller M (2008) Regulation of transporters by nuclear hormone receptors: implications during inflammation. Mol Pharm 5: 67-76. https://doi.org/10.1021/mp700102q
89. Yu C, Argyropoulos G, Zhang Y, et al. (2008) Neuroinflammation activates Mdr1b efflux transport through NFkappaB: promoter analysis in BBB endothelia. Cell Physiol Biochem 22: 745-56. https://doi.org/10.1159/000185558
90. Pan W, Yu C, Hsuchou H, et al. (2010) The role of cerebral vascular NFkappaB in LPS-induced inflammation: differential regulation of efflux transporter and transporting cytokine receptors. Cell Physiol Biochem 25: 623-30. https://doi.org/10.1159/000315081
91. Kosters A, Karpen SJ (2010) The role of inflammation in cholestasis: clinical and basic aspects. Semin Liver Dis 30:186-94. https://doi.org/10.1055/s-0030-1253227
92. Tirona RG (2011) Molecular mechanisms of drug transporter regulation. Handb Exp Pharmacol 2011: 373-402. https://doi.org/10.1007/978-3-642-14541-4_10
93. Harvey RD, Morgan ET (2014) Cancer, inflammation, and therapy: effects on cytochrome p450-mediated drug metabolism and implications for novel immunotherapeutic agents. Clin Pharmacol Ther 96: 449-57. https://doi.org/10.1038/clpt.2014.143
94. Morgan ET (1997) Regulation of cytochromes P450 during inflammation and infection. Drug Metab Rev 29: 1129-88. https://doi.org/10.3109/03602539709002246
95. Aitken AE, Morgan ET (2007) Gene-specific effects of inflammatory cytokines on cytochrome P450 2C, 2B6 and 3A4 mRNA levels in human hepatocytes. Drug Metab Dispos 35: 1687-93. https://doi.org/10.1124/dmd.107.015511
96. Muntane-Relat J, Ourlin JC, Domergue J, et al. (1995) Differential effects of cytokines on the inducible expression of CYP1A1, CYP1A2, and CYP3A4 in human hepatocytes in primary culture. Hepatology 22: 1143-53.
97. Calleja C, C Eeckhoutte, Dacasto M, et al. (1998) Comparative effects of cytokines on constitutive and inducible expression of the gene encoding for the cytochrome P450 3A6 isoenzyme in cultured rabbit hepatocytes: consequences on progesterone 6beta-hydroxylation. Biochem Pharmacol 56: 1279-85. https://doi.org/10.1016/s0006-2952(98)00178-6
98. Tapner M, Liddle C, Goodwin B, et al. (1996) Interferon gamma down-regulates cytochrome P450 3A genes in primary cultures of well-differentiated rat hepatocytes. Hepatology 24: 367-73. https://doi.org/10.1002/hep.510240213
99. Monshouwer M, Witkamp RF, Nujmeijer SM, et al. (1996) Suppression of cytochrome P450- and UDP glucuronosyl transferase-dependent enzyme activities by proinflammatory cytokines and possible role of nitric oxide in primary cultures of pig hepatocytes. Toxicol Appl Pharmacol 137: 237-44. https://doi.org/10.1006/taap.1996.0077
100. Rubin K, Janefeldt A, Andersson L, et al. (2015) HepaRG cells as human-relevant in vitro model to study the effects of inflammatory stimuli on cytochrome P450 isoenzymes. Drug Metab Dispos 43: 119-25. https://doi.org/10.1124/dmd.114.059246
101. Abdel-Razzak Z, Loyer P, Fautrel A, et al. (1993) Cytokines down-regulate expression of major cytochrome P-450 enzymes in adult human hepatocytes in primary culture. Mol Pharmacol 44: 707-15.
102. Pan J, Xiang Q, Ball S (2000) Use of a novel real-time quantitative reverse transcription-polymerase chain reaction method to study the effects of cytokines on cytochrome P450 mRNA expression in mouse liver. Drug Metab Dispos 28: 709-13.
103. Morgan ET (1993) Down-regulation of multiple cytochrome P450 gene products by inflammatory mediators in vivo. Independence from the hypothalamo-pituitary axis. Biochem Pharmacol 45: 415-9. https://doi.org/10.1016/0006-2952(93)90078-b
104. Frye RF, Schneider VM, Frye CS, et al. (2002) Plasma levels of TNF-alpha and IL-6 are inversely related to cytochrome P450-dependent drug metabolism in patients with congestive heart failure. J Card Fail 8: 315-9. https://doi.org/10.1054/jcaf.2002.127773
105. Chaluvadi MR, Kinloch RD, Nyagode BA, et al. (2009) Regulation of hepatic cytochrome P450 expression in mice with intestinal or systemic infections of citrobacter rodentium. Drug Metab Dispos 37: 366-74. https://doi.org/10.1124/dmd.108.024240
106. Masubuchi Y, Enoki K, Horie T (2008) Down-regulation of hepatic cytochrome P450 enzymes in rats with trinitrobenzene sulfonic acid-induced colitis. Drug Metab Dispos 36: 597-603. https://doi.org/10.1124/dmd.107.018754
107. Dickmann LJ, McBride HJ, Patel SK, et al. (2012) Murine collagen antibody induced arthritis (CAIA) and primary mouse hepatocyte culture as models to study cytochrome P450 suppression. Biochem Pharmacol 83: 1682-9. https://doi.org/10.1016/j.bcp.2012.03.001
108. Sanada H, Sekimoto M, Kamoshita A, et al. (2011) Changes in expression of hepatic cytochrome P450 subfamily enzymes during development of adjuvant-induced arthritis in rats. J Toxicol Sci 36: 181-90. https://doi.org/10.2131/jts.36.181
109. Ashino T, Arima Y, Shioda S, et al. (2007) Effect of interleukin-6 neutralization on CYP3A11 and metallothionein-1/2 expressions in arthritic mouse liver. Eur J Pharmacol 558: 199-207. https://doi.org/10.1016/j.ejphar.2006.11.072
110. Ling S, Jamali F (2005) Effect of early phase adjuvant arthritis on hepatic P450 enzymes and pharmacokinetics of verapamil: an alternative approach to the use of an animal model of inflammation for pharmacokinetic studies. Drug Metab Dispos 33: 579-86. https://doi.org/10.1124/dmd.104.002360
111. Dickmann LJ, Patel SK, Wienkers LC, et al. (2012) Effects of interleukin 1beta (IL-1beta) and IL-1beta/interleukin 6 (IL-6) combinations on drug metabolizing enzymes in human hepatocyte culture. Curr Drug Metab 13: 930-7. https://doi.org/10.2174/138920012802138642
112. Iber H, Q Chen, P Y Cheng, et al. (2000) Suppression of CYP2C11 gene transcription by interleukin-1 mediated by NF-kappaB binding at the transcription start site. Arch Biochem Biophys 377: 187-94. https://doi.org/10.1006/abbi.2000.1772
113. Parmentier JH, Kremers P, Ferrari L, et al. (1993) Repression of cytochrome P450 by cytokines: IL-1 beta counteracts clofibric acid induction of CYP4A in cultured fetal rat hepatocytes. Cell Biol Toxicol 9: 307-13. https://doi.org/10.1007/bf00755608
114. Bleau AM, Levitchi MC, Maurice H, et al. (2000) Cytochrome P450 inactivation by serum from humans with a viral infection and serum from rabbits with a turpentine-induced inflammation: the role of cytokines. Br J Pharmacol, 130: 1777-84. https://doi.org/10.1038/sj.bjp.0703486
115. Donato MT, Guillén MI, Jover R, et al. (1997) Nitric oxide-mediated inhibition of cytochrome P450 by interferon-gamma in human hepatocytes. J Pharmacol Exp Ther 281: 484-90.
116. Nadin L, Butler AM, Farrell GC, et al. (1995) Pretranslational down-regulation of cytochromes P450 2C11 and 3A2 in male rat liver by tumor necrosis factor alpha. Gastroenterology 109: 198-205. https://doi.org/10.1016/0016-5085(95)90285-6
117. Gorski JC, Hall SD, Becker P, et al. (2000) In vivo effects of interleukin-10 on human cytochrome P450 activity. Clin Pharmacol Ther 67: 32-43. https://doi.org/10.1067/mcp.2000.103860
118. Elkahwaji J, Robin MA, Berson A, et al. (1999) Decrease in hepatic cytochrome P450 after interleukin-2 immunotherapy. Biochem Pharmacol 57: 951-4. https://doi.org/10.1016/s0006-2952(98)00372-4
119. Tinel M, Robin MA, Doostzadeh J, et al. (1995) The interleukin-2 receptor down-regulates the expression of cytochrome P450 in cultured rat hepatocytes. Gastroenterology 109: 1589-99. https://doi.org/10.1016/0016-5085(95)90648-7
120. Yang J, Hao C, Yang D, et al. (2010) Pregnane X receptor is required for interleukin-6-mediated down-regulation of cytochrome P450 3A4 in human hepatocytes. Toxicol Lett 197: 219-26. https://doi.org/10.1016/j.toxlet.2010.06.003
121. Dickmann LJ, Patel SK, Rock DA, et al. (2011) Effects of interleukin-6 (IL-6) and an anti-IL-6 monoclonal antibody on drug-metabolizing enzymes in human hepatocyte culture. Drug Metab Dispos 39: 1415-22. https://doi.org/10.1124/dmd.111.038679
122. Nyagode BA, Lee CM, Morgan ET (2010) Modulation of hepatic cytochrome P450s by Citrobacter rodentium infection in interleukin-6- and interferon-{gamma}-null mice. J Pharmacol Exp Ther 335: 480-8. http://dx.doi.org/10.1124/jpet.110.171488
123. Williams JF, Bement WJ, Sinclair JF, et al. (1991) Effect of interleukin 6 on phenobarbital induction of cytochrome P-450IIB in cultured rat hepatocytes. Biochem Biophys Res Commun 178: 1049-55. https://doi.org/10.1016/0006-291x(91)90998-m
124. Chen YL, Florentin I, Batt AM, et al. (1992) Effects of interleukin-6 on cytochrome P450-dependent mixed-function oxidases in the rat. Biochem Pharmacol 44: 137-48. https://doi.org/10.1016/0006-2952(92)90047-m
125. Siewert E, Bort R, Kluge R, et al. (2000) Hepatic cytochrome P450 down-regulation during aseptic inflammation in the mouse is interleukin 6 dependent. Hepatology 32: 49-55. https://doi.org/10.1053/jhep.2000.8532
126. Morgan ET (1989) Suppression of constitutive cytochrome P-450 gene expression in livers of rats undergoing an acute phase response to endotoxin. Mol Pharmacol 36: 699-707.
127. Robertson GR, Field J, Goodwin B, et al. (2003) Transgenic mouse models of human CYP3A4 gene regulation. Mol Pharmacol 64: 42-50. https://doi.org/10.1124/mol.64.1.42
128. Kacevska M, Mahns A, Sharma R, et al. (2013) Extra-hepatic cancer represses hepatic drug metabolism via interleukin (IL)-6 signalling. Pharm Res 30: 2270-8. https://doi.org/10.1007/s11095-013-1042-3
129. Mimura H, Kobayashi K, Xu L, et al. (2015) Effects of cytokines on CYP3A4 expression and reversal of the effects by anti-cytokine agents in the three-dimensionally cultured human hepatoma cell line FLC-4. Drug Metab Pharmacokinet 30: 105-10. https://doi.org/10.1016/j.dmpk.2014.09.004
130. Nyagode BA, Jahangardi R, Merrell MD, et al. (2014) Selective effects of a therapeutic protein targeting tumor necrosis factor-alpha on cytochrome P450 regulation during infectious colitis: Implications for disease-dependent drug-drug interactions. Pharmacol Res Perspect 2: e00027. https://doi.org/10.1002/prp2.27
131. Kinloch RD, Lee CM, Rooijen NV, et al. (2011) Selective role for tumor necrosis factor-alpha, but not interleukin-1 or Kupffer cells, in down-regulation of CYP3A11 and CYP3A25 in livers of mice infected with a noninvasive intestinal pathogen. Biochem Pharmacol 82: 312-21. https://doi.org/10.1016/j.bcp.2011.04.016
132. Ling S, Jamali F (2009) The effect of infliximab on hepatic cytochrome P450 and pharmacokinetics of verapamil in rats with pre-adjuvant arthritis: a drug-disease and drug-drug interaction. Basic Clin Pharmacol Toxicol 105: 24-9. https://doi.org/10.1111/j.1742-7843.2009.00405.x
133. Lee CM, Pohl J, Morgan ET (2009) Dual mechanisms of CYP3A protein regulation by proinflammatory cytokine stimulation in primary hepatocyte cultures. Drug Metab Dispos 37: 865-72. https://doi.org/10.1124/dmd.108.026187
134. Carlson TJ, Billings RE (1996) Role of nitric oxide in the cytokine-mediated regulation of cytochrome P-450. Mol Pharmacol 49: 796-801.
135. Mimche SM, Lee CM, Liu KH, et al. (2019) A non-lethal malarial infection results in reduced drug metabolizing enzyme expression and drug clearance in mice. Malar J 18: 234. https://dx.doi.org/10.1186%2Fs12936-019-2860-5
136. Nguyen TV, Ukairo O, Khetani SR, et al. (2015) Establishment of a hepatocyte-kupffer cell coculture model for assessment of proinflammatory cytokine effects on metabolizing enzymes and drug transporters. Drug Metab Dispos 43: 774-85. https://doi.org/10.1124/dmd.114.061317
137. Hartmann G, Cheung AK, Piquette-Miller M (2002) Inflammatory cytokines, but not bile acids, regulate expression of murine hepatic anion transporters in endotoxemia. J Pharmacol Exp Ther 303: 273-81. https://doi.org/10.1124/jpet.102.039404
138. Klein M, Thomas M, Hofmann U, et al. (2015) A systematic comparison of the impact of inflammatory signaling on absorption, distribution, metabolism, and excretion gene expression and activity in primary human hepatocytes and HepaRG cells. Drug Metab Dispos 43: 273-83. https://doi.org/10.1124/dmd.114.060962
139. Mimche SM, Nyagode BA, Merrell MD, et al. (2014) Hepatic cytochrome P450s, phase II enzymes and nuclear receptors are downregulated in a Th2 environment during Schistosoma mansoni infection. Drug Metab Dispos 42: 134-40. https://doi.org/10.1124/dmd.113.054957
140. Yang K (2020) What do we know about remdesivir drug interactions? Clin Transl Sci 13: 842-844. https://doi.org/10.1111/cts.12815
141. Mishima E, Anzai N, Miyazaki M, et al. (2020) Uric acid elevation by favipiravir, an antiviral drug. Tohoku J Exp Med 251: 87-90. https://doi.org/10.1620/tjem.251.87
142. Karbanova S, L Cerveny, L Jiraskova, et al. (2019) Transport of ribavirin across the rat and human placental barrier: Roles of nucleoside and ATP-binding cassette drug efflux transporters. Biochem Pharmacol 163: 60-70. https://doi.org/10.1016/j.bcp.2019.01.024
143. Fukuchi Y, Furihata T, Hashizume M, et al. (2010) Characterization of ribavirin uptake systems in human hepatocytes. J Hepatol 52: 486-92. https://doi.org/10.1016/j.jhep.2010.01.011
144. Chen C, Han YH, Yang Z, et al. (2011) Effect of interferon-alpha2b on the expression of various drug-metabolizing enzymes and transporters in co-cultures of freshly prepared human primary hepatocytes. Xenobiotica 41: 476-85. https://doi.org/10.3109/00498254.2011.560971
145. Kiser JJ, Gerber JG, Predhomme JA, et al. (2008) Drug/Drug interaction between lopinavir/ritonavir and rosuvastatin in healthy volunteers. J Acquir Immune Defic Syndr 47: 570-8. https://doi.org/10.1097/qai.0b013e318160a542
146. Van Waterschoot RA, Ter Heine R, Wagenaar E, et al. (2010) Effects of cytochrome P450 3A (CYP3A) and the drug transporters P-glycoprotein (MDR1/ABCB1) and MRP2 (ABCC2) on the pharmacokinetics of lopinavir. Br J Pharmacol 160: 1224-33. https://doi.org/10.1111/j.1476-5381.2010.00759.x
147. Rakhmanina NY, Neely MN, Schaik RHNV, et al. (2011) CYP3A5, ABCB1, and SLCO1B1 polymorphisms and pharmacokinetics and virologic outcome of lopinavir/ritonavir in HIV-infected children. Ther Drug Monit 33: 417-24. https://doi.org/10.1097/ftd.0b013e318225384f
148. Gupta A, Zhang Y, Unadkat JD, et al. (2004) HIV protease inhibitors are inhibitors but not substrates of the human breast cancer resistance protein (BCRP/ABCG2). J Pharmacol Exp Ther 310: 334-41. https://doi.org/10.1124/jpet.104.065342
149. Janneh O, Jones E, Chandler B, et al. (2007) Inhibition of P-glycoprotein and multidrug resistance-associated proteins modulates the intracellular concentration of lopinavir in cultured CD4 T cells and primary human lymphocytes. J Antimicrob Chemother 60: 987-93. https://doi.org/10.1093/jac/dkm353
150. Storch CH, Theile D, Lindenmaier H, et al., (2007) Comparison of the inhibitory activity of anti-HIV drugs on P-glycoprotein. Biochem Pharmacol 73: 1573-81. https://doi.org/10.1016/j.bcp.2007.01.027
151. Weiss J, Rose J, Storch CH, et al. (2007) Modulation of human BCRP (ABCG2) activity by anti-HIV drugs. J Antimicrob Chemother 59: 238-45. https://doi.org/10.1093/jac/dkl474
152. Marzolini C, Gibbons S, Khoo S, et al. (2016) Cobicistat versus ritonavir boosting and differences in the drug-drug interaction profiles with co-medications. J Antimicrob Chemother 71: 1755-8. https://doi.org/10.1093/jac/dkw032
153. Cvetkovic M, Leake B, Fromm MF, et al. (1999) OATP and P-glycoprotein transporters mediate the cellular uptake and excretion of fexofenadine. Drug Metab Dispos 27: 866-71.
154. Zhang L, Gorset W, Washington CB, et al. (2000) Interactions of HIV protease inhibitors with a human organic cation transporter in a mammalian expression system. Drug Metab Dispos 28: 329-34.
155. van der Sandt IC, Vos CM, Nabulsi L, et al. (2001) Assessment of active transport of HIV protease inhibitors in various cell lines and the in vitro blood--brain barrier. AIDS 15: 483-91. https://doi.org/10.1097/00002030-200103090-00007
156. Huisman MT, Smit JW, Crommentuyn KML, et al. (2002) Multidrug resistance protein 2 (MRP2) transports HIV protease inhibitors, and transport can be enhanced by other drugs. AIDS 16: 2295-301. https://doi.org/10.1097/00002030-200211220-00009
157. Perloff MD, von Moltke LL, Greenblatt DJ (2002) Greenblatt, Fexofenadine transport in Caco-2 cells: inhibition with verapamil and ritonavir. J Clin Pharmacol 42: 1269-74. https://doi.org/10.1177/009127002762491370
158. Tirona RG, Leake BF, Wolkoff AW, et al. (2003) Human organic anion transporting polypeptide-C (SLC21A6) is a major determinant of rifampin-mediated pregnane X receptor activation. J Pharmacol Exp Ther 304: 223-8. https://doi.org/10.1124/jpet.102.043026
159. Jung N, Lehmann C, Rubbert A, et al. (2008) Relevance of the organic cation transporters 1 and 2 for antiretroviral drug therapy in human immunodeficiency virus infection. Drug Metab Dispos 36: 1616-23. https://doi.org/10.1124/dmd.108.020826
160. Xu C, Zhu L, Chan T, et al. (2016) Chloroquine and hydroxychloroquine are novel inhibitors of human organic anion transporting polypeptide 1A2. J Pharm Sci 105: 884-890. https://doi.org/10.1002/jps.24663
161. Sortica VA, Lindenau JD, Cunha MG, et al. (2017) SLCO1A2, SLCO1B1 and SLCO2B1 polymorphisms influences chloroquine and primaquine treatment in Plasmodium vivax malaria. Pharmacogenomics 18: 1393-1400. https://doi.org/10.2217/pgs-2017-0077
162. Weiss JG, Bajraktari-Sylejmani G, Haefeli WE (2020) Interaction of hydroxychloroquine with pharmacokinetically important drug transporters. Pharmaceutics 12:919. https://doi.org/10.3390/pharmaceutics12100919
163. Courtois A, Payen L, Guillouzo A, et al. (1999) Up-regulation of multidrug resistance-associated protein 2 (MRP2) expression in rat hepatocytes by dexamethasone. FEBS Lett 459: 381-5. https://doi.org/10.1016/s0014-5793(99)01295-8
164. Manceau S, Giraud C, Declèves X, et al. (2012) Expression and induction by dexamethasone of ABC transporters and nuclear receptors in a human T-lymphocyte cell line. J Chemother 24: 48-55. https://doi.org/10.1179/1120009x12z.00000000010
165. Lespine A, Dupuy J, Orlowski S, et al. (2006) Interaction of ivermectin with multidrug resistance proteins (MRP1, 2 and 3). Chem Biol Interact 159: 169-79. https://doi.org/10.1016/j.cbi.2005.11.002
166. Jani M, Makai I, Kis E, et al. (2011) Ivermectin interacts with human ABCG2. J Pharm Sci 100: 94-7. https://doi.org/10.1002/jps.22262
167. Houshaymi B, Nasreddine N, Kedees M, et al. (2019) Oleic acid increases uptake and decreases the P-gp-mediated efflux of the veterinary anthelmintic Ivermectin. Drug Res (Stuttg) 69: 173-180. https://doi.org/10.1055/a-0662-5741
168. Perloff ES, Duan SX, Skolnik PR, et al. (2005) Atazanavir: effects on P-glycoprotein transport and CYP3A metabolism in vitro. Drug Metab Dispos 33: 764-70. https://doi.org/10.1124/dmd.104.002931
169. Kis O, Zastre JA, Hoque MT, et al. (2013) Role of drug efflux and uptake transporters in atazanavir intestinal permeability and drug-drug interactions. Pharm Res 30: 1050-64. https://doi.org/10.1007/s11095-012-0942-y
170. Bousquet L, Roucairol C, Hembury A, et al. (2008) Comparison of ABC transporter modulation by atazanavir in lymphocytes and human brain endothelial cells: ABC transporters are involved in the atazanavir-limited passage across an in vitro human model of the blood-brain barrier. AIDS Res Hum Retroviruses 24: 1147-54. https://doi.org/10.1089/aid.2007.0022
171. Zastre JA, Chan GNY, Ronaldson PT, et al. (2009) Up-regulation of P-glycoprotein by HIV protease inhibitors in a human brain microvessel endothelial cell line. J Neurosci Res 87: 1023-36. https://doi.org/10.1002/jnr.21898
172. Sugie M, Asakura E, Zhao YL, et al. (2004) Possible involvement of the drug transporters P glycoprotein and multidrug resistance-associated protein Mrp2 in disposition of azithromycin. Antimicrob Agents Chemother 48: 809-14. https://doi.org/10.1128/aac.48.3.809-814.2004
173. Lan T, Rao A, Haywood J, et al. (2009) Interaction of macrolide antibiotics with intestinally expressed human and rat organic anion-transporting polypeptides. Drug Metab Dispos 37: 2375-82. https://doi.org/10.1124/dmd.109.028522
174. Holmstock N, Mols R, Annaert P, et al. (2010) In situ intestinal perfusion in knockout mice demonstrates inhibition of intestinal p-glycoprotein by ritonavir causing increased darunavir absorption. Drug Metab Dispos 38: 1407-10. https://doi.org/10.1124/dmd.110.032771
175. Tong L, Phan TK, Robinson KL, et al. (2007) Effects of human immunodeficiency virus protease inhibitors on the intestinal absorption of tenofovir disoproxil fumarate in vitro. Antimicrob Agents Chemother 51: 3498-504. https://doi.org/10.1128/aac.00671-07
176. Brown KC, Paul S, Kashuba ADM (2009) Drug interactions with new and investigational antiretrovirals. Clin Pharmacokinet 48: 211-41. https://doi.org/10.2165/00003088-200948040-00001
177. Fujimoto H, Higuchi M, Watanabe H, et al. (2009) P-glycoprotein mediates efflux transport of darunavir in human intestinal Caco-2 and ABCB1 gene-transfected renal LLC-PK1 cell lines. Biol Pharm Bull 32: 1588-93. https://doi.org/10.1248/bpb.32.1588
178. Vermeir M, Lachau-Durand S, Mannens G, et al. (2009) Absorption, metabolism, and excretion of darunavir, a new protease inhibitor, administered alone and with low-dose ritonavir in healthy subjects. Drug Metab Dispos 37: 809-20. https://doi.org/10.1124/dmd.108.024109
179. Holmstock N, Gonzalez FJ, Baes M, et al. (2013) PXR/CYP3A4-humanized mice for studying drug-drug interactions involving intestinal P-glycoprotein. Mol Pharm 10: 1056-62. https://doi.org/10.1021/mp300512r
180. Posada MM, Cannady EA, Payne CD, et al. (2017) Prediction of transporter-mediated drug-drug interactions for baricitinib. Clin Transl Sci 10: 509-519. https://doi.org/10.1111/cts.12486
181. Hodge C, Marra F, Marzolini C, et al. (2020) Drug interactions: a review of the unseen danger of experimental COVID-19 therapies. J Antimicrob Chemother 75: 3417-3424. https://doi.org/10.1093/jac/dkaa340
182. Yamakawa Y, Hamada A, Nakashima R, et al. (2011) Association of genetic polymorphisms in the influx transporter SLCO1B3 and the efflux transporter ABCB1 with imatinib pharmacokinetics in patients with chronic myeloid leukemia. Ther Drug Monit 33: 244-50. https://doi.org/10.1097/ftd.0b013e31820beb02
183. Mlejnek P, Kosztyu P, Dolezel P, et al. (2017) Reversal of ABCB1 mediated efflux by imatinib and nilotinib in cells expressing various transporter levels. Chem Biol Interact 273: 171-179. https://doi.org/10.1016/j.cbi.2017.06.012
184. Alsherbiny MA, Li CG (2018) Medicinal cannabis-potential drug interactions. Medicines (Basel) 6: 3. https://doi.org/10.3390/medicines6010003
185. Garrison KL, Garrison KL, German P, et al. (2018) The drug-drug interaction potential of antiviral agents for the treatment of chronic Hepatitis C infection. Drug Metab Dispos 46: 1212-1225. https://doi.org/10.1124/dmd.117.079038
186. Garimella T, You X, Wang R, et al. (2016) A review of daclatasvir drug-drug interactions. Adv Ther 33: 1867-1884. https://doi.org/10.1007/s12325-016-0407-5
187. Du YX, Chen XP (2020) Favipiravir: Pharmacokinetics and concerns about clinical trials for 2019-nCoV infection. Clin Pharmacol Ther 108: 242-247. https://doi.org/10.1002/cpt.1844
188. Becquemont L, Chazouilleres O, Serfaty L, et al. (2002) Effect of interferon alpha-ribavirin bitherapy on cytochrome P450 1A2 and 2D6 and N-acetyltransferase-2 activities in patients with chronic active hepatitis C. Clin Pharmacol Ther 71: 488-95. https://doi.org/10.1067/mcp.2002.124468
189. Aungst BJ, Nguyen NH, Bulgarelli JP, et al. (2000) The influence of donor and reservoir additives on Caco-2 permeability and secretory transport of HIV protease inhibitors and other lipophilic compounds. Pharm Res 17: 1175-80. https://doi.org/10.1023/a:1026402410783
190. Elewa H, Wilby KJ (2017) A review of pharmacogenetics of antimalarials and associated clinical implications. Eur J Drug Metab Pharmacokinet 42: 745-756. https://doi.org/10.1007/s13318-016-0399-1
191. Babayeva M, Loewy Z (2020) Repurposing drugs for COVID-19: Pharmacokinetics and pharmacogenomics of chloroquine and hydroxychloroquine. Pharmgenomics Pers Med 13: 531-542. https://doi.org/10.2147/PGPM.S275964
192. Lee JY, Vinayagamoorthy N, Han K, et al. (2016) Association of polymorphisms of cytochrome P450 2D6 with blood hydroxychloroquine levels in patients with systemic lupus erythematosus. Arthritis Rheumatol 68: 184-90. https://doi.org/10.1002/art.39402
193. Pascussi JM, Drocourt L, Gerbal-Chaloin S, et al. (2001) Dual effect of dexamethasone on CYP3A4 gene expression in human hepatocytes. Sequential role of glucocorticoid receptor and pregnane X receptor. Eur J Biochem 268: 6346-58. https://doi.org/10.1046/j.0014-2956.2001.02540.x
194. Deng P, Zhong D, Yu K, et al. (2013) Pharmacokinetics, metabolism, and excretion of the antiviral drug arbidol in humans. Antimicrob Agents Chemother 57: 1743-55. https://doi.org/10.1128/aac.02282-12
195. Bernareggi A, Borghi A, Borgonovi M, et al. (1992) Teicoplanin metabolism in humans. Antimicrob Agents Chemother 36: 1744-9. https://doi.org/10.1128/aac.36.8.1744
196. Broekhuysen J, Stockis A, Lins RL, et al. (2000) Nitazoxanide: pharmacokinetics and metabolism in man. Int J Clin Pharmacol Ther 38: 387-94. https://doi.org/10.5414/cpp38387
197. Neodo A, Schulz JD, Huwyler J, et al. (2019) In Vitro and In Vivo drug-drug interaction study of the effects of ivermectin and oxantel pamoate on tribendimidine. Antimicrob Agents Chemother 63: e00762-18. https://doi.org/10.1128/aac.00762-18
198. Westphal JF (2000) Macrolide - induced clinically relevant drug interactions with cytochrome P-450A (CYP) 3A4: an update focused on clarithromycin, azithromycin and dirithromycin. Br J Clin Pharmacol 50: 285-95. https://doi.org/10.1046/j.1365-2125.2000.00261.x
199. Galetin A, Burt H, Gibbons L, et al. (2006) Prediction of time-dependent CYP3A4 drug-drug interactions: impact of enzyme degradation, parallel elimination pathways, and intestinal inhibition. Drug Metab Dispos 34: 166-75. https://doi.org/10.1124/dmd.105.006874
200. Febvre-James M, Bruyère A, Vée ML, et al. (2018) The JAK1/2 inhibitor ruxolitinib reverses interleukin-6-mediated suppression of drug-detoxifying proteins in cultured human hepatocytes. Drug Metab Dispos 46: 131-140. https://doi.org/10.1124/dmd.117.078048
201. Aslanis V, Umehara K, Huth F, et al. (2019) Multiple administrations of fluconazole increase plasma exposure to ruxolitinib in healthy adult subjects. Cancer Chemother Pharmacol 84: 749-757. https://doi.org/10.1007/s00280-019-03907-1
202. Takahashi T, Luzum JA, Nicol MR, et al. (2020) Pharmacogenomics of COVID-19 therapies. NPJ Genom Med 5: 35. https://doi.org/10.1038/s41525-020-00143-y
203. Gschwind HP, Pfaar U, Waldmeier F, et al. (2005) Metabolism and disposition of imatinib mesylate in healthy volunteers. Drug Metab Dispos 33: 1503-12. https://doi.org/10.1124/dmd.105.004283
204. Filppula AM, Neuvonen M, Laitila J, et al. (2013) Autoinhibition of CYP3A4 leads to important role of CYP2C8 in imatinib metabolism: variability in CYP2C8 activity may alter plasma concentrations and response. Drug Metab Dispos 41: 50-9. https://doi.org/10.1124/dmd.112.048017
205. Kashuba AD, Nafziger AN, Kearns GL, et al. (1998) Effect of fluvoxamine therapy on the activities of CYP1A2, CYP2D6, and CYP3A as determined by phenotyping. Clin Pharmacol Ther 64: 257-68. https://doi.org/10.1016/s0009-9236(98)90174-6
206. Iga K (2016) Dynamic and static simulations of fluvoxamine-perpetrated drug-drug interactions using multiple cytochrome P450 inhibition modeling, and determination of perpetrator-specific CYP isoform inhibition constants and fractional CYP isoform contributions to victim clearance. J Pharm Sci 105: 1307-17. https://doi.org/10.1016/j.xphs.2015.11.044
207. Christensen M, Tybring G, Mihara K, et al. (2002) Low daily 10-mg and 20-mg doses of fluvoxamine inhibit the metabolism of both caffeine (cytochrome P4501A2) and omeprazole (cytochrome P4502C19). Clin Pharmacol Ther 71: 141-52. https://doi.org/10.1067/mcp.2002.121788
208. Stout SM, Cimino NM (2014) Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev 46: 86-95. https://doi.org/10.3109/03602532.2013.849268
209. Kirby BJ, Symonds WT, Kearney BP, et al. (2015) Pharmacokinetic, pharmacodynamic, and drug-interaction profile of the Hepatitis C virus NS5B polymerase inhibitor sofosbuvir. Clin Pharmacokinet 54: 677-90. https://doi.org/10.1007/s40262-015-0261-7
210. Tempestilli M, Caputi P, Avataneo V, et al. (2020) Pharmacokinetics of remdesivir and GS-441524 in two critically ill patients who recovered from COVID-19. J Antimicrob Chemother 75: 2977-2980. https://doi.org/10.1093/jac/dkaa239
211. Humeniuk R, Mathias A, Cao H, et al. (2020) Safety, tolerability, and pharmacokinetics of remdesivir, an antiviral for treatment of COVID-19, in healthy subjects. Clin Transl Sci 13: 896-906. https://doi.org/10.1111/cts.12840
212. Humeniuk R, Mathias A, Kirby BJ, et al. (2021) Pharmacokinetic, pharmacodynamic, and drug-interaction profile of remdesivir, a SARS-CoV-2 replication inhibitor. Clin Pharmacokinet 60: 569-583. https://doi.org/10.1007/s40262-021-00984-5
213. Projean D, Baune B, Farinotti R, et al. (2003) In vitro metabolism of chloroquine: identification of CYP2C8, CYP3A4, and CYP2D6 as the main isoforms catalyzing N-desethylchloroquine formation. Drug Metab Dispos 31: 748-54. https://doi.org/10.1124/dmd.31.6.748
214. Furst DE (1996) Pharmacokinetics of hydroxychloroquine and chloroquine during treatment of rheumatic diseases. Lupus 5 Suppl 1: S11-5.
215. Paniri A, Hosseinia MM, Rasoulinejad A, et al. (2020) Molecular effects and retinopathy induced by hydroxychloroquine during SARS-CoV-2 therapy: Role of CYP450 isoforms and epigenetic modulations. Eur J Pharmacol 886: 173454. https://doi.org/10.1016/j.ejphar.2020.173454
216. Arshad S, Kilgore P, Chaudhry ZS, et al. (2020) Treatment with hydroxychloroquine, azithromycin, and combination in patients hospitalized with COVID-19. Int J Infect Dis 97: 396-403. https://doi.org/10.1016/j.ijid.2020.06.099
217. Lane JCE, Weaver J, Kostka K, et al. (2020) Risk of hydroxychloroquine alone and in combination with azithromycin in the treatment of rheumatoid arthritis: a multinational, retrospective study. Lancet Rheumatol 2: e698-e711. https://doi.org/10.1016/s2665-9913(20)30276-9
218. Jiao T, Yao X, Zhao Y, et al. (2020) Dexamethasone-induced liver enlargement is related to PXR/YAP activation and lipid accumulation but not hepatocyte proliferation. Drug Metab Dispos 48: 830-839. https://doi.org/10.1124/dmd.120.000061
219. Buckley DB, Klaassen CD (2009) Induction of mouse UDP-glucuronosyltransferase mRNA expression in liver and intestine by activators of aryl-hydrocarbon receptor, constitutive androstane receptor, pregnane X receptor, peroxisome proliferator-activated receptor alpha, and nuclear factor erythroid 2-related factor 2. Drug Metab Dispos 37: 847-56. https://doi.org/10.1124/dmd.108.024190
220. Cheng X, Maher J, Dieter MZ, et al. (2005) Regulation of mouse organic anion-transporting polypeptides (Oatps) in liver by prototypical microsomal enzyme inducers that activate distinct transcription factor pathways. Drug Metab Dispos 33: 1276-82. https://doi.org/10.1124/dmd.105.003988
221. Hartley DP, Dai X, He YD, et al. (2004) Activators of the rat pregnane X receptor differentially modulate hepatic and intestinal gene expression. Mol Pharmacol 65: 1159-71. https://doi.org/10.1124/mol.65.5.1159
222. Wang H, Faucette SR, Gilbert D, et al. (2003) Glucocorticoid receptor enhancement of pregnane X receptor-mediated CYP2B6 regulation in primary human hepatocytes. Drug Metab Dispos 31: 620-30. https://doi.org/10.1124/dmd.31.5.620
223. Kliewer SA (2003) The nuclear pregnane X receptor regulates xenobiotic detoxification. J Nutr 133: 2444S-2447S. https://doi.org/10.1093/jn/133.7.2444S
224. Song QQ, Xie WY, Tang YJ, et al. (2017) Genetic variation in the glucocorticoid pathway involved in interindividual differences in the glucocorticoid treatment. Pharmacogenomics 18: 293-316. https://doi.org/10.2217/pgs-2016-0151
225. Dunn ST, Hedges L, Sampson KE, et al. (2011) Pharmacokinetic interaction of the antiparasitic agents ivermectin and spinosad in dogs. Drug Metab Dispos 39: 789-95. https://doi.org/10.1124/dmd.110.034827
226. Ballent M, Lifschitz A, Virkel G, et al. (2007) Involvement of P-glycoprotein on ivermectin kinetic behaviour in sheep: itraconazole-mediated changes on gastrointestinal disposition. J Vet Pharmacol Ther 30: 242-8. https://doi.org/10.1111/j.1365-2885.2007.00848.x
227. Ballent M, Lifschitz A, Virkel G, et al. (2006) Modulation of the P-glycoprotein-mediated intestinal secretion of ivermectin: in vitro and in vivo assessments. Drug Metab Dispos 34: 457-63. https://doi.org/10.1124/dmd.105.007757
228. Real R, Egido E, Pérez M, et al. (2011) Involvement of breast cancer resistance protein (BCRP/ABCG2) in the secretion of danofloxacin into milk: interaction with ivermectin. J Vet Pharmacol Ther 34: 313-21. https://doi.org/10.1111/j.1365-2885.2010.01241.x
229. Schmith VD, Zhou JJ, Lohmer LRL (2020) The Approved Dose of Ivermectin Alone is not the Ideal Dose for the Treatment of COVID-19. Clin Pharmacol Ther 108: 762-765. https://doi.org/10.1002/cpt.1889
230. Umehara K, Huth F, Jin Y, et al. (2019) Drug-drug interaction (DDI) assessments of ruxolitinib, a dual substrate of CYP3A4 and CYP2C9, using a verified physiologically based pharmacokinetic (PBPK) model to support regulatory submissions. Drug Metab Pers Ther 34. https://doi.org/10.1515/dmpt-2018-0042
231. Filppula AM, Laitila J, Neuvonen PJ, et al. (2012) Potent mechanism-based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br J Pharmacol 165: 2787-98. https://doi.org/10.1111/j.1476-5381.2011.01732.x
232. Breedveld P, Pluim D, Cipriani G, et al. (2005) The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res 65: 2577-82. https://doi.org/10.1158/0008-5472.can-04-2416
233. Burger H, Tol HV, Boersma AWM, et al. (2004) Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 104: 2940-2. https://doi.org/10.1182/blood-2004-04-1398
234. von Mehren M, Widmer N (2011) Correlations between imatinib pharmacokinetics, pharmacodynamics, adherence, and clinical response in advanced metastatic gastrointestinal stromal tumor (GIST): an emerging role for drug blood level testing? Cancer Treat Rev 37: 291-9. https://doi.org/10.1016/j.ctrv.2010.10.001
235. Picard S, Titier K, Etienne G, et al. (2007) Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood 109: 3496-9. https://doi.org/10.1182/blood-2006-07-036012
236. Vignali C, Moretti M, Quaiotti J, et al. (2020) Distribution of fluvoxamine and identification of the main metabolite in a fatal intoxication. J Anal Toxicol 45: e1-e5. https://doi.org/10.1093/jat/bkaa084
237. Obach RS, Ryder TF (2010) Metabolism of ramelteon in human liver microsomes and correlation with the effect of fluvoxamine on ramelteon pharmacokinetics. Drug Metab Dispos 38: 1381-91. https://doi.org/10.1124/dmd.110.034009
238. Kanacher T, Lindauer A, Mezzalana E, et al. (2020) A physiologically-based pharmacokinetic (PBPK) model network for the prediction of CYP1A2 and CYP2C19 drug-drug-gene interactions with fluvoxamine, omeprazole, S-mephenytoin, moclobemide, tizanidine, mexiletine, ethinylestradiol, and caffeine. Pharmaceutics 12: 1191. https://doi.org/10.3390/pharmaceutics12121191