Commentary on: p53 G242A Mutant Expression by Cancer Cells Averts Immune Assault by Natural Killer Cells
Authors: Ronald A. Hill, Saqline Mostaq, Amanda Raphael, Yong-Yu Liu*
School of Basic Pharmaceutical and Toxicological Sciences, University of Louisiana at Monroe, Monroe, Louisiana, USA
*Correspondence to: Professor. Yong-Yu Liu, School of Basic Pharmaceutical and Toxicological Sciences, University of Louisiana at Monroe, 700 University Avenue, Monroe, Louisiana 71209, USA; E-mail: yliu@ulm.edu
Received: 01 November 2022; Accepted: 21 November 2022; Published: 28 November 2022
Copyright: © 2022 Hill RA. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation: Hill RA, Mostaq S, Raphael A, Liu YY (2022) Commentary on: p53 G242A Mutant Expression by Cancer Cells Averts Immune Assault by Natural Killer Cells, 21st Century Pathology, Volume 2 (6): 134
Abstract
p53 mutant presence can protect cancerous cells from immunity; however, underlying mechanisms remained to be identified. In a recent Exp Cell Res article, researchers report that EMT6 breast cancer cells (wild-type Trp53) evoked NK cell proliferation in mice spleens, whereas SVTneg2 cells (Trp53 G242A+/+) suppressed NK cell production. In co-culture, EMT6 cells caused NK cell proliferation, markedly increasing interferon-gamma (IFN-γ) production; however, SVTneg2 cells repressed NK cell responses. Additionally, p53 G242A presence in cancer cells promoted Mult-1 expression with suppressed H60a expression (activating or inhibitory ligands to NKG2D receptors) subverting NK cells, and enabling these cancer cells to escape immune destruction.
Keywords:
p53 tumor suppressor; Natural killer cell; Missense mutation; Breast cancer
Introduction
Natural killer (NK) cells play critical roles in immune surveillance and destruction. NK cells recognize cancerous cells expressing low levels of major histocompatibility complex (MHC) class I molecules, and kill cells by secreting cytokines, including interferon-gamma (IFN-γ) and tumor necrosis factor (TNF) [1]. These processes are mediated through the interactions of receptors on NK cells with complementary ligands expressed on cancerous cells [2]. Natural killer group member 2D (NKG2D) is one of the most important stimulatory receptors on NK cells [3]. The family of NKG2D ligands include MHC class I-chain-related proteins A (MICA) and B (MICB), retinoic acid early transcript 1 (Rae1), histocompatibility antigen 60 (H60), and mouse UL16-binding protein-like transcript 1 (Mult1) [3]. These ligands are minimally expressed by normal cells, but are often induced on cancer cells [4]. Suppressing expression of these stimulatory ligands that can be recognized by and activate NKG2D receptors is one means by which cancerous cells escape antitumor immunity. Tumor suppressor p53 acts as an essential transcription factor for expressing p53-responsive genes. The TP53 gene is somatically mutated in over 50% of all cancer cases, and more than 75% of these alterations are missense mutants (mm) [5, 6]. Four hotspot mutation sites (R175, G245, R248, R273 codons) are prominently associated with unfavorable prognosis in breast cancer [7, 8]. In a recent study, we, for the first time, elucidated how a p53 G245A mutant enabled cancerous cells to avert cytotoxic assault by NK cells, thereby evading immune eradication [9].
Materials and Methods
Murine breast carcinoma SVTneg2 and EMT6 cell lines carry Trp53 G242A+/+ mutation (corresponding to human hot-spot mutation G245A+/+) and wt Trp53, respectively [8,10]. Orthotopic breast tumors generated from SVTneg2 or EMT6 cells were transplanted into the fat pads of mice (wt Trp53), after which tumor growth and rejection were followed. Splenic production of NK cells (NKGC2+) and cytotoxic T lymphocytes (CD8+) by mice inoculated (tail vein) with cancer cells of SVTneg2 or EMT6 lines was assessed using imaging flow cytometry (IFC). In parallel NK cells were co-cultured with SVTneg2 or EMT6 cells to assess activation of NK cells and evoked production of INF-γ. Methods and experimental conditions are further detailed in [9].
Results
p53 mutant fostered tumor growth and impeded regression of tumors in mice
To explore whether p53 expression and functions connect to host immune rejection and tumor regression, we firstly implanted punch samples from resected primary tumors into mice carrying wild-type p53 (WT). SVTneg2 (Trp53 G242A+/+) tumors grew faster and lasted longer than EMT6 tumors, although tumors of either origin greatly regressed or disappeared by day 24 after implantation (Figure 1A). Average tumor volumes of SVTneg2 tumors on days 8 and 10 were significantly greater than for EMT6 tumors, respectively, by approximately 2-fold (p<0.01). SVTneg2 tumors grew larger and persisted longer during the regression phase in host mice, as assessed by volume AUC8→24d, which was approximately 68% greater than for EMT6 tumors (p>0.01) (Figure 1B).
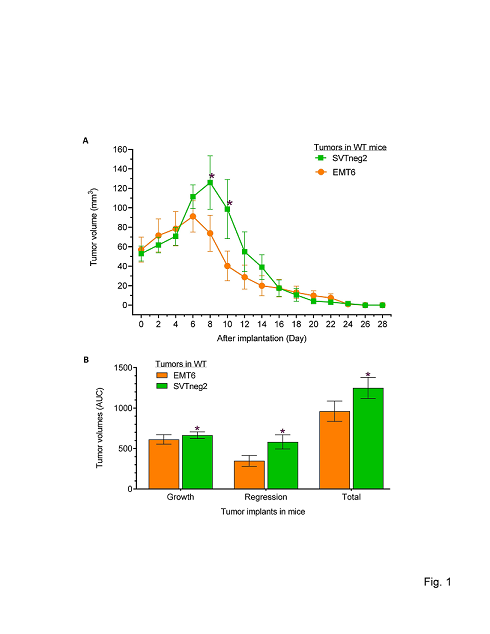
Figure 1: p53 mutant fostered tumor growth and impeded regression of breast cancer in mice. (A) Tumor growth and hindered regression of tumors in mice (12 cases/group). Punches from primary breast tumors generated from EMT6 and SVTneg2 cell lines were transplanted (3.5 mm3/side) into mammary fat pads of mice (EMT6 left side, SVTneg2 right side, 12 cases/group). Multiple comparisons were carried out via two-way ANOVA with Bonferroni’s post-hoc test. *, p<0.01 compared to EMT6 tumors in WT mice; &, p<0.01 compared to SVTneg2 tumors in WT mice. (B) Areas under the curve (AUC) for tumor growth were assessed from Days 0-8 as baseline in WT mice. AUCs for tumor regression were assessed from Days 8-24 and baseline for EMT6 tumor explants, and Days 10-26 for SVTneg2 explants. Total AUC values account for tumor growth and regression over the entire study period. *, p<0.01 compared to EMT6 tumors in WT mice.
p53 mutant-carrying cancer cells reduced host NK cell numbers, whereas wild-type cells increased NK cell populations
To understand how p53 mediates anticancer immunity associated with tumor regression, we injected cancer cells as well as normal fibroblasts and saline as control into tail veins of mice (WT Trp53), and assessed NK cells (NKG2D+) and cytotoxic T lymphocytes (CD8+) using imaging flow cytometry (IFC). We found that EMT6 cancer cells and fibroblasts significantly increased the numbers of NK and CD8+ T cells, as compared to a saline-treated group (Figures 2C, 2D). Interestingly, injection of SVTneg2 cancer cells elicited substantially lesser increases in NK cell numbers, as compared with responses to EMT6 cancer cells or normal fibroblasts, by more than 2-fold (Figure 2D). However, SVTneg2 cells did significantly increase the numbers of CD8+ T cells, by greater than 2-fold (802 cells vs. 267 or 300 cells), as compared to either EMT6 cancer cells or fibroblasts (Figure 2D).
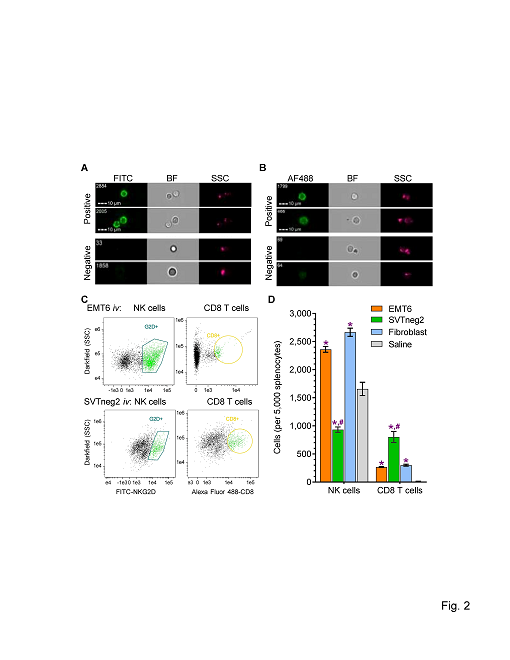
Figure 2: Stimulating effects of p53 in breast cancer cells on host NK cells and CD8+ T cells. Cells (3 x 106 cells/mouse) of EMT6 (wt-p53) or SVTneg2 (mm-p53 G242A) breast cancer lines and normal fibroblasts (wt-p53) were administrated into tail veins of mice (WT Trp53, once every 3 days (on days 1, 4, 7, and 10; 5 cases/group). Spleens were dissected on day 10, and isolated splenocytes were analyzed using imaging IFC following incubation with antibodies for epitope detection. (A) Representative images of NKG2D+ NK cells and NKG2D negative cells. NK cells were characterized by FITC-NKG2D+ (FITC green), compared with bright field (BF) and side-scatter (SSC). It was estimated that the NKG2D+ splenic cells included approximately 3% of γδ T cells and αβCD8+ T cells [21]. (B) Representative images of CD8+ T cells and negative cells. Alexa Fluor488-CD8+ (AF488 green), compared with bright field (BF) and side-scatter (SSC). (C) Populations of NK cells and CD8 T cells. G2D+, NKG2D+. (D) SVTneg2 cancer cells decreased NK cells and increased CD8+ T cells among mouse splenocytes at the end of the series of cell injections. Multiple comparisons were carried out via one-way ANOVA with Tukey’s post-hoc test. *, p<0.001 compared to saline; #, p<0.001 compared to EMT6 or fibroblasts.
p53 mutant presence in cancer cells repressed NK cell activation
We directly examined the effects of p53 expression by cancer cells, and mm-p53 vs. wt-p53, on activation of NK cells (Trp53) in co-culture. As compared to fibroblasts, EMT6 cells (wt-p53) significantly increased NK cell presence (assessed as overall viability), whereas SVTneg2 cells (mm-p53) repressed the viability (Figure 3A). Further, the magnitudes of the effects on the NK cell viability were dependent on the number of cancer cells added. To wit, increasing EMT6 cells (500 to 2,000 cells) incubated with NK cells (at 20,000/well) elicited additional increases in NK cells, respectively, as compared to the same added numbers of normal fibroblasts. In contrast, increasing numbers of SVTneg2 cells consistently decreased viable NK cells, by approximately 10-fold, 6-fold and 4-fold, respectively (Figure 3A). Furthermore, silencing wt-p53 expression by EMT-6 cells with siRNA-p53 significantly decreased viable NK cells in co-culture, while silencing mm-p53 expression by SVTneg2 cells increased NK cell numbers (Figure 3A). These effects of p53 on NK cells were verified with microscopy of co-cultured cells (Figure 3B).
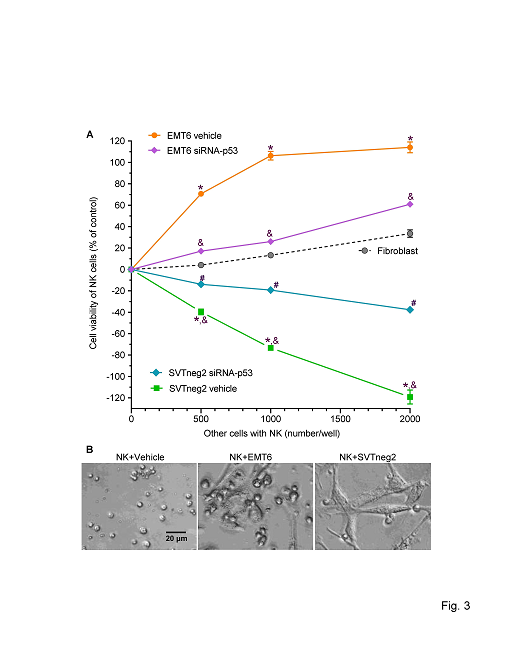
Figure 3: p53 in breast cancer cells stimulates NK cell proliferation in co-culture. Isolated NK cells (20,000/well in 96-well plates) were co-cultured with breast cancer cells (0, 500, 1000, 2000) of the EMT6 (wt-p53) line, or of the SVTneg2 (mm-p53) line, or with normal fibroblasts, in 10% FBS medium for 48 h. Cells were transfected with 100 nM of siRNA-p53 (siRNA-p53) or siRNA-scramble (Vehicle) before co-culture. (A) p53 G242A suppressed NK cell viability and caused death of NK cells. Multiple comparisons were carried out via two-way ANOVA with Bonferroni’s post-hoc test. *, p>0.001 compared to NK cells co-cultured with fibroblasts; &, p<0.001 compared to NK cells co-cultured with EMT6 vehicle; #, p<0.01 compared with NK cells co-cultured with SVTneg2 vehicle. (B) Photomicrographs of NK cells with cancer cells in co-culture. NK cells (20,000 cells/well) were cultured with EMT6 (1000 cells) or SVTneg2 (1000 cells) in 10% FBS DMEM medium, and observed and processed with the EVOS FL cell imaging system with color CCD camera (200x magnification).
p53 of cancer cells modulated their expression of cell-surface NKG2D ligands to increase NK cell IFN-γ production
Interferon-γ (IFN-γ) is a pleiotropic cytokine, produced mainly by NK cells. We assessed levels of IFN-γ released into the media during co-culture with cancer cells. We found that IFN-γ levels from NK co-culture with SVTneg2 cells were approximately 5-fold lower than for co-culture with EMT6 cells (Figure 4A). Concordantly, silencing p53 expression in EMT6 cells with siRNA-p53 decreased IFN-γ levels, by approximately 2-fold, in co-culture with NK cells. However, similar treatments with siRNA-p53 that silenced mm-p53 expression in SVTneg2 cells elevated the IFN-γ levels, by more than 3-fold (4.3 vs. 1.2, p<0.001), as compared to co-culture with non-silenced SVTneg2 cells (Figure 4B).
To characterize how p53 of cancer cells impacted NK cell activation, we further assessed the effects of NKG2D ligand expression by cancer cells. In conjunction with production of phosphorylated p53 (pp53), EMT6 cancer cells expressed substantial levels of Mult-1, but relatively lower levels of H60a (Figures 4B, 4C). SVTneg2 cells exhibited substantially lower levels of pp53, and also of Mult-1, approximately 10-fold and 3-fold lower, respectively, whereas H60a levels were elevated 1.6-fold. Furthermore, silencing p53 expression in SVTneg2 cells with siRNA-p53 significantly increased Mult-1 expression over levels seen in non-silenced cells, whereas H60a levels were seen to decrease significantly (Figures 4B, 4C).
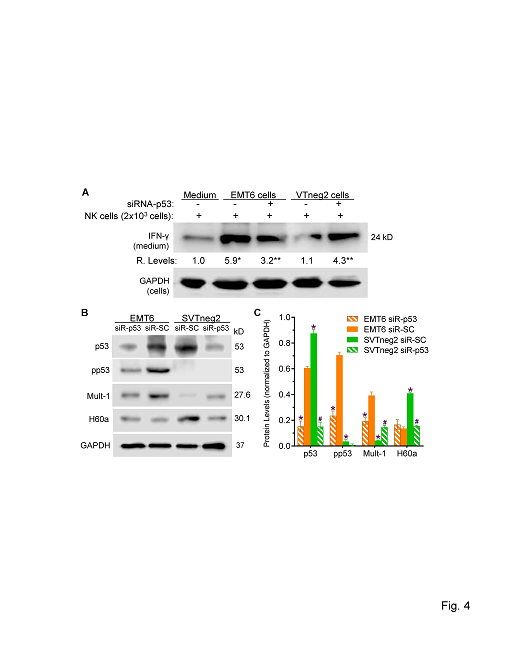
Figure 4: Effects of p53 on expression of NKG2D ligands by breast cancer cells and on INF-γ production by NK cells. (A) wt-p53 expression by cancer cells activated NK cells to release INF-γ. NK cells (20,000 cells/well) were co-cultured in 10% FBS medium for 48 h with cells (2000 cells; 6 wells per condition) of EMT6 or SVTneg2 lines. siRNA-p53 treatments (100 nM, 48 h) silenced the expression of p53 in cells. Multiple comparisons were carried out via one-way ANOVA with Tukey’s post-hoc test. *, p<0.001 compared to NK cells alone; **, p<0.001 compared to NK cells cultured with EMT6 or SVTneg2 cells treated with scrambled control (siRNA-SC). (B) Western blots of p53 and NKG2D ligands expressed by cancer cells. EMT6 or SVTneg2 cancer cells were treated with siRNA-p53 or siRNA-SC (100 nM) for 48 h. Equal amounts of soluble proteins (50 μg/lane) were resolved on SDS-PAGE and immunoblotted with corresponding antibodies. (C) mm-p53 (G242A+/+) presence altered the protein expression of Mult-1 and of H60a in cancer cells. Protein levels are presented as density values of each protein, normalized against GAPDH, from three different blot analyses. Multiple comparisons were carried out via one-way ANOVA with Tukey’s post-hoc test. *, p<0.001 compared to EMT6 siRNA-SC (EMT6 siR-SC); #, p<0.001 compared to SVTneg2 siRNA-SC (SVTneg2 siR-SC).
Altogether, these data indicate that Mult1 expressed on EMT6 cells (wt Trp53) activates NKG2D receptors, causing NK propagation with elevated IFN-γ production. By contrast, up-regulated expression of H60a and down-regulated Mult-1 expression on SVTneg2 cells (Trp53 G242A) deactivates NK cells and decreases IFN-γ production. Consequently, wt p53 presence enhances immune surveillance and destruction against cancer cells, whereas mutant p53 allows cancer cells to escape immune surveillance and eradication, enabling cancer progression, as diagrammed in Figure 5.
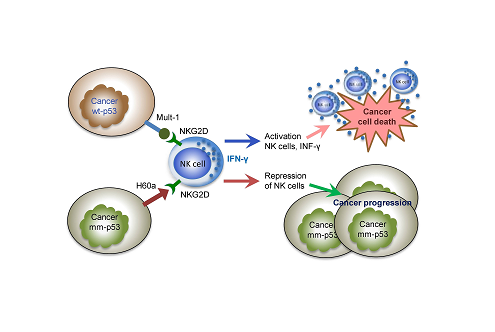
Figure 5: p53 modulates expression of NKG2D ligands by cancer cells to activate NK cells and eradicate tumors. Normal response mediated by the actions of p53 protein (wild-type) enhance the expression of stimulating ligands including Mult-1 in breast cancer cells, thereby activating NK cells via NKG2D receptors to increase NK cell proliferation and IFN-γ production, thus enabling the immune-eradication of cancer cells carrying wt-p53. Missense p53 mutant protein presence increases the expression of inhibitory NKG2D ligand H60a, and inhibits expression of activating NKG2D ligand Mult1, thereby repressing NK cell activation and proliferation, in turn abrogating immune-eradication of cancerous cells, resulting in cancer progression.
Discussion
This study, for the first time, elucidated that marked mutant p53 (G242A+/+) could suppress anticancer immunity via deactivation of NK cells to enable and accelerate cancer progression. Prominent wt-p53 expression by cancer cells augments immune surveillance, enabling successful detection and eradication of cancer cells through processes heavily reliant on NK cell recruitment and activation, as seen here with the example of EMT6 tumors implanted in WT mice (Figure 1A). Although mutation hotspots, including G245, account for approximately 30% of p53 mutations in human breast cancers [7], it has been unclear what roles extant mm-p53 expression in cancers play in subverting anticancer immunity. In our study, mouse SVTneg2 tumors expressing a mm-p53 (G242A+/+, corresponding to human G245A+/+) grew significantly larger and regressed more slowly in WT mice than did EMT6 tumors expressing wt-p53. Consistent with the findings described above, SVTneg2 cancer cells substantially suppressed propagation of NK cells in mice systemically injected with these cells, and concordantly, inhibited the proliferation and induced the death of NK cells in co-culture (cf. Figures 2D, 3A).
Our studies further characterized that p53 modulates the expression of NKG2D ligands on cancer cells to activate NK cells for proliferation and IFN-γ production. The capability of NK cells to attack cancer cells has been identified in many studies, and that has been further proven to rely on wt-p53 of cancer cells [11-13]. Concordantly, we found that while EMT6 cells directly activated NK cells in co-culture, and elicited higher levels of IFN-γ release into the medium (cf. Figures 3A, 4A, 4B), by contrast, interestingly, mutant p53 of cancer cells (SVTneg2 carrying Trp53 G242A+/+) directly suppressed the activation of NK cells and reduced IFN-γ levels in co-culture. These findings, together with those noted above, clearly highlight and further reaffirm that NK cells and IFN-γ are prominent components of the p53-activated immune surveillance assault against cancer cells, which may at least contribute to the onset of adaptive immune defense, helping bring about rejection of growing and developing tumors. Our results also show that while mm-p53 cancer cells (exemplified by SVTneg2 cells) may retain the ability to stimulate host mouse (WT) generation of CD8+ T cells, such compromised immune responses are likely insufficient to prevent tumor progression, at least per models exploited for the present studies.
Previous studies have shown that tumors transplanted into experimental animals are generally rejected by immune responses involving NK cells, attributable to the expression in tumor cells of NKG2D ligands that activate natural-killer group 2D (NKG2D) receptors on NK cell surfaces to activate and propagate NK cells [14, 15]. Various signaling mechanisms, including ones involving E2F transcription factor, PI3K, Ras oncogene activation, and DNA damage response, have been reported to induce production of NKG2D ligands [16-18]. Activation of p53-mediated functions has also been reported to stimulate the expression of the NKG2D ligand ULBP2 in human cancer cells [19]. Similarly, the down-regulation or suppression of activating ligands for NKG2D receptors on NK cells can help cancer cells avoid immune surveillance and destruction [15, 20]. Our assessments of altered NK cell responses and tumor rejection concordantly affirmed key Mult1 and H60a roles in p53-driven regulation of NK cell responses to murine breast cancer cells.
NK cells can swiftly kill multiple neighboring cells if these show surface markers associated with oncogenic transformation. This unique property, and the resultant capacity of NK cells to enhance antibody-mediated and T cell-enacted responses, poise NK cells to provide a key component of anticancer immunity. TP53 missense mutation is among the most common of genetic alterations detected in cancers, including breast cancers. To reiterate, our study demonstrates that missense p53 G242A can suppress spleen NK cell responses in mice, thus substantially subverting antitumor immune surveillance. Further studies will be required to investigate whether or not other hotspots of TP53 mutation act similarly in suppressing NK cell activation to subvert anticancer immunity. Due to limitations imposed by available samples, we were unable to analyze the alterations of NK cells or CD8+ T cells in tumors of mice after transplantation, or in the peripheral blood of mice after injections of tumor cells. NK cells and other immune cells in spleen are able to respond directly to injected tumor cells, and newly generated immune cells immediately distribute to tissues, including peripheral blood.
Conclusion
Mutant p53 (mm, G242A, corresponding to human hot-spot mutation G245A) expression in cancer cells, by acting to repress expression of activating ligands to NKG2D receptors, subverts NK cell immune responses, thus defecting anticancer immunity. Further studies are required to investigate whether other mutation hotspots of TP53 act similarly, and to explore possible involvement of other immune cell types and extent thereof.
Conflict of Interest
YY Liu is a member of the Scientific Advisory Board for Sanofi-Genzyme; no other authors have competing interests.
References
1. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. science. 2011 Jan 7;331(6013):44-9. https://doi.org/10.1126/science.1198687
2. Raulet DH, Guerra N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nature Reviews Immunology. 2009 Aug;9(8):568-80. https://doi.org/10.1038/nri2604
3. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nature Reviews Immunology. 2003 Oct;3(10):781-90. https://doi.org/10.1038/nri1199
4. Ni J, Miller M, Stojanovic A, Cerwenka A. Toward the next generation of NK cell-based adoptive cancer immunotherapy. Oncoimmunology. 2013 Apr 1;2(4):e23811. https://doi.org/10.4161/onci.23811
5. Leroy B, Anderson M, Soussi T. TP 53 mutations in human cancer: database reassessment and prospects for the next decade. Human mutation. 2014 Jun;35(6):672-88. https://doi.org/10.1002/humu.22552
6. Liu YY. Resuscitating Wild-Type p53 Expression by Disrupting Ceramide Glycosylation: A Novel Approach to Target Mutant p53 TumorsResuscitation of Wild-Type p53 Expression by Ceramide. Cancer research. 2011 Oct 15;71(20):6295-9. https://doi.org/10.1158/0008-5472.CAN-11-0700
7. Walerych D, Napoli M, Collavin L, Del Sal G. The rebel angel: mutant p53 as the driving oncogene in breast cancer. Carcinogenesis. 2012 Nov 1;33(11):2007-17. https://doi.org/10.1093/carcin/bgs232
8. Graessmann M, Berg B, Fuchs B, Klein A, Graessmann A. Chemotherapy resistance of mouse WAP-SVT/t breast cancer cells is mediated by osteopontin, inhibiting apoptosis downstream of caspase-3. Oncogene. 2007 May;26(20):2840-50. https://doi.org/10.1038/sj.onc.1210096
9. Uddin MB, Roy KR, Hill RA, Roy SC, Gu X, Li L, Zhang QJ, You Z, Liu YY. p53 missense mutant G242A subverts natural killer cells in sheltering mouse breast cancer cells against immune rejection. Experimental Cell Research. 2022 May 18:113210. https://doi.org/10.1016/j.yexcr.2022.113210
10. Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clinical Cancer Research. 2005 Dec 15;11(24):8782-8. https://doi.org/10.1158/1078-0432.CCR-05-1664
11. Mahdi T, Tanzer J, Brizard A, Guilhot F, Babin P, Kitzis A. Cell-mediated cytotoxicity can be regulated by p53 tumor suppressor gene activity in vitro. Biology of the Cell. 1995 Jan 1;84(3):175-85. https://doi.org/10.1016/0248-4900(96)89427-5
12. Chollat-Namy M, Ben Safta-Saadoun T, Haferssas D, Meurice G, Chouaib S, Thiery J. The pharmalogical reactivation of p53 function improves breast tumor cell lysis by granzyme B and NK cells through induction of autophagy. Cell death & disease. 2019 Sep 20;10(10):1-20. https://doi.org/10.1038/s41419-019-1950-1
13. Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. Journal of Experimental Medicine. 2013 Sep 23;210(10):2057-69. https://doi.org/10.1084/jem.20130783
14. Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nature immunology. 2000 Aug;1(2):119-26. https://doi.org/10.1038/77793
15. Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, Xu J, Rovis TL, Xiong N, Raulet DH. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science. 2015 Apr 3;348(6230):136-9. https://doi.org/10.1126/science.1258867
16. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005 Aug;436(7054):1186-90. https://doi.org/10.1038/nature03884
17. Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. Journal of Experimental Medicine. 2012 Dec 17;209(13):2409-22. https://doi.org/10.1084/jem.20120565
18. Tokuyama M, Lorin C, Delebecque F, Jung H, Raulet DH, Coscoy L. Expression of the RAE-1 family of stimulatory NK-cell ligands requires activation of the PI3K pathway during viral infection and transformation. PLoS pathogens. 2011 Sep 22;7(9):e1002265. https://doi.org/10.1371/journal.ppat.1002265
19. Textor S, Fiegler N, Arnold A, Porgador A, Hofmann TG, Cerwenka A. Human NK Cells Are Alerted to Induction of p53 in Cancer Cells by Upregulation of the NKG2D Ligands ULBP1 and ULBP2ULBP1 and ULBP2 Are Direct p53 Target Genes. Cancer research. 2011 Sep 15;71(18):5998-6009. https://doi.org/10.1158/0008-5472.CAN-10-3211
20. Sheppard S, Ferry A, Guedes J, Guerra N. The paradoxical role of NKG2D in cancer immunity. Frontiers in immunology. 2018:1808. https://doi.org/10.3389/fimmu.2018.01808
21. Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity. 2002 Jul 1;17(1):19-29. https://doi.org/10.1016/S1074-7613(02)00333-3