An Annotation on Axonal Degeneration in Very Early Guillain-Barré Syndrome
Author: Jose Berciano*
Professor emeritus ad honorem, University of Cantabria, University Hospital ?Marqu
Citation: Citation: Berciano J (2021) An Annotation on Axonal Degeneration in Very Early Guillain-Barré Syndrome, 21st Century Pathol, Volume 1 (1): 106
Abstract
This is a short review analysing pathological events in very early Guillain-Barré syndrome (VEGBS; ≤ 4 days after onset). At this stage, conventional nerve conduction studies allow disease dichotomization into demyelinating and axonal forms in a minority of cases. Pathological studies of both classic VEGBS and experimental autoimmune neuritis have demonstrated that the main lesion is inflammatory edema involving proximal nerve trunks, particularly spinal nerves when neither demyelination nor axonal degeneration has entered into the scene. Such edema would be pathogenic using the increase of endoneurial fluid pressure in nerve trunks possessing epi-perineurium, which constricts transperineurial microcirculation causing ischemic nerve damage. All these notions have pathophysiological, diagnostic, and therapeutic implications.
Keywords:
Acute inflammatory demyelinating polyneuropathy; Acute motor axonal neuropathy; Axonal degeneration; Demyelination; Endoneurial fluid pressure; Endoneurial ischemia; Inflammatory oedema; Methylprednisolone; Nerve conduction studies; Perineurium; Spinal nerves; Spinal roots; Ultrasonography; Very early Guillain-Barré syndrome
Guillain-Barre Syndrome: Basic Notions
Guillain-Barre syndrome (GBS) is an acute-onset, immune-mediated disorder of the peripheral nervous system, which includes at least three disease patterns: acute inflammatory demyelinating polyneuropathy (AIDP), acute motor axonal and motor-sensory axonal neuropathy (AMAN and AMSAN), and Miller Fisher syndrome [1,2]. Furthermore, GBS is sub-classified into classical (weakness involving all four limbs) and localized subtypes [3]. Experimental autoimmune neuritis (EAN) is a widely accepted model of GBS [4]. Anti-ganglioside antibodies in AMAN/AMSAN are complement-fixing that mainly bind to GM1 and GD1a gangliosides; in animal models, they induce axonal damage by fixing complement, recruiting macrophages, and depositing membrane attack complex in the axolemma membrane [2]. By contrast with AMAN, the immunological cascade involved in AIDP is less understood, given that specific antibody biomarkers have yet to be characterized.
By definition, maximum weakness is reached within 4 weeks after symptom onset, but most patients have already reached their maximum weakness within 2 weeks [2].
Nerve conduction studies in very early GBS stages
Nerve conduction studies play an important role in GBS diagnosis and classification [5]. Nevertheless, in very early stages of classic GBS (VEGBS; ? 4 days after onset) disease dichotomization into demyelinating and axonal electrophysiological forms, may only be possible in around 20% of cases [6,7]; intriguingly, this is a diagnostic constraint occurring even in patients with severe muscle weakness that require mechanical ventilation. One may well wonder how to explain such clinical-pathological discrepancy. To resolve this dilemma, a clear notion of the inaugural pathological background, both in VEGBS and EAN, is essential [8].
Topography of Initial GBS Lesions
Theoretically, the bulk of early inflammatory edema should predominate in nerve segments with the less efficient blood-nerve barrier, including spinal roots, spinal ganglia, spinal nerves, and pre-terminal and terminal nerve segments [8]. Autopsy studies in classic VEGBS have demonstrated that, indeed, the main lesion is inflammatory edema involving proximal nerve trunks, when neither axonal degeneration nor demyelination has entered into the scene [8-10]. Furthermore, initial edema is more prominent where motor and sensory roots join to form the spinal nerve. As an approximation, (figure 1) illustrates this pathological hallmark in an early, classic GBS patient (11). It is worth noting that in their original pathological description of AMAN, McKhann GM, et al. (1993) found extensive Wallerian-like degeneration of the ventral roots and, usually, of motor fibers within peripheral nerves; the proportion of degenerating radicular fibers increased distally toward the ventral root exit from the dura where 80% of fibers were degenerating [12], namely maximal pathology also occurred here in spinal nerves. Given that such topography is unexpected in a primary axonopathy, we have proposed that in very early AMAN there may be a dual mechanism of axonal damage [8]: i/ ganglioside-mediated nodal/paranodal conduction block of motor nerves implying axonal dysfunction, and eventually Wallerian-like degeneration; and ii/ conduction block at ventral rami of spinal nerves caused by endoneurial ischemia inducing axonal damage (vide infra).
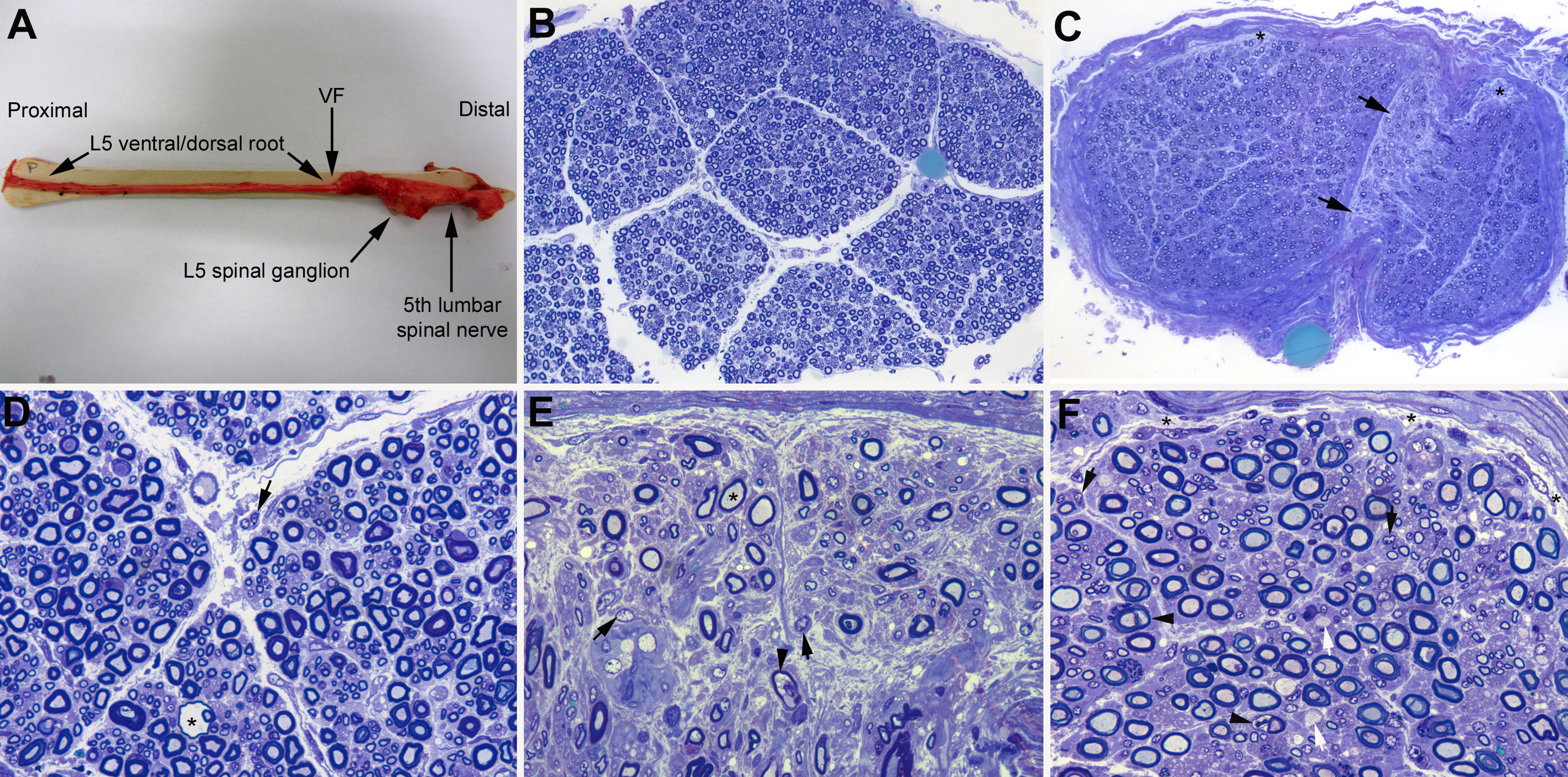
Figure 1: Pathological features in L5 spinal root, fifth lumbar nerve and sciatic nerve (adapted from case 1 by Gallardo and colleagues [11]). In short, woman aged 80 years with a 4-day history of ascending quadriplegia requiring mechanical ventilation (for electrophysiological and sonographic findings, see reference [11]). Clinical diagnosis was GBS. She died on day 9 after onset. (A) After being dissected down, macroscopic appearance of the right L5 spinal root, L5 spinal ganglion and fifth lumbar spinal nerve. Whereas the pre-foraminal root shows normal morphology, as of the vertebral foramen (VF) note visible nerve enlargement. (B) Semithin cross-section of L5 ventral root, taken 1 cm above its entrance to the VF, showing that the density of myelinated fibers is preserved; note the absence of epi- perineurium (Toluidine blue; original magnification x100 before reduction). (C) Semithin cross-section of the ventral ramus of the fifth lumbar nerve, taken at its emergence trough intervertebral foramen, showing widespread endoneurial oedema, which is more conspicuous in septum adjacent areas (arrows) and sub-perineurial areas (asterisks); such oedema results in a spacing out phenomenon giving an observer the false impression of reduced density of myelinated fibers (Toluidine blue; original magnification x65 before reduction). (D) High-power view of the L5 ventral root showing preservation of the density of myelinated fibres with occasional presence of mononuclear cells (arrow) and a fibre exhibiting myelin vacuolization (asterisk). (E) High-power view of the sub-septum area arrowed in C. Note the presence of florid inflammatory oedema with numerous mononuclear cells (arrows), fibres with inappropriately thin myelin sheaths (asterisk), and fibres exhibiting myelin vacuolization (arrowhead). Having in mind the spacing out phenomenon, there is an apparent reduced density of myelin fibres in comparison with L5 ventral root and sciatic nerve (previous and next images) (Toluidine blue; original magnification x630 before reduction). (F) Semithin section of sciatic nerve showing some demyelinated axons (white arrows), fibres with vacuolar degeneration (arrowheads), and widespread but slight endoneurial oedema more marked in sub-perineurial areas (asterisks) with presence of mononuclear cells (black arrows); as consequence of this the spacing out phenomenon is barely perceptible (Toluidine blue; original magnification x630 before reduction). Summarizing, the outstanding lesion was endoneurial inflammatory oedema predominating in ventral ramus of the fifth lumbar nerve.
The time sequence of lesions in T cell-induced P2-EAN in Lewis rats was masterfully described by Izumo and colleagues [13]: i/ the disease starts with flaccid tail and hind-limb paralysis between days 3.5 and 4 of post-inoculation (pi) with autoreactive T cell lines sensitized to residue 57-81 of P2 myelin protein; ii/ the first pathological change comes up on day 4 pi, consisting of epi-endoneurial inflammatory edema; and iii/ between days 7 and 9 pi, appearance of demyelination and axonal degeneration. Such inflammatory edema is pathogenic using the increase of endoneurial fluid pressure (EFP) in nerve trunks possessing epi-perineurium, which constricts transperineurial microcirculation producing rapid ischemic nerve injury [14]. Intriguingly, areas of endoneurial ischemia have been reported in fatal AIDP cases [15,16]. Although so exact chronology of EAN pathological events is not necessarily applicable to GBS, it helps to interpret its early pathophysiology. Edema of spinal nerves in VEGBS, either AIDP or AMAN/AMSAN, could manifest with proximal conduction failure causing tetra paresis, even when conventional nerve conduction studies are normal or minimally altered; its electrophysiological counterpart would be abnormal late responses (F-wave, H reflex) [6,7]. Some special electrophysiological studies, such as lumbar root stimulation or triple stimulation technique, have demonstrated selective conduction failure in proximal nerve trunks in early AIDP [17] or AMAN [18], respectively. Nerve ultra-sonographic studies have corroborated that spinal nerve edema is a hotspot in the early stages of any GBS subtype, either demyelinating or axonal [6,11].
Microscopic Anatomy of the Peripheral Nervous System: An Essential Concept
Knowledge of the microscopic anatomy of the peripheral nervous system is essential for an adequate understanding of the pathogenic relevance of early pathological events in GBS [19,20] (figure 2). Spinal roots traverse the subarachnoid space covered by an elastic multicellular root sheath derived from the arachnoid and penetrate the dura at the subarachnoid angle. As of the subarachnoid angle, where motor and sensory roots join to form the spinal nerve, the dura mater is in continuity with the epineurium, whereas the arachnoid turns into perineurium. Therefore, intrathecal nerve roots are covered by an elastic root sheath, whereas spinal nerves and more distant nerve trunks till their pre-terminal segments possess epi-perineurium that is relatively inelastic. Conceivably, initial inflammatory edema may be accommodated in intrathecal nerve roots enlarging their size but without this implying a significant increase of EFP. Conversely, in nerve trunks surrounded by epi-perineurium, such edema may cause a pathogenic elevation of EFP, which leads to ischemic conduction failure, and eventually to Wallerian-like degeneration [20] (vide supra and figure 2). Although this phenomenon may occur in any segment of peripheral nerve trunks, pathological and ultrasonographic studies indicate that spinal nerves are the hotspot in any early GBS subtype, thus explaining the high prevalence of electrophysiological changes pointing to pathology in proximal nerve segments. In any case, inflammatory edema is also a histological feature of intermediate and pre-terminal nerve segments, a potential cause of partial conduction block, nerve inexcitability, or reversible conduction failure [6-8,20,21].

Figure 2: Diagram of spinal root and spinal nerve microscopic anatomy (taken from Berciano and colleagues [20]). As of subarachnoid angle (SA), the epineurium (Ep) is in continuity with the dura mater (DM). The endoneurium (En) persists from the peripheral nerves through the spinal roots to their junction with the spinal cord. At the SA, the greater portion of the perineurium (Pe) passes between the dura and the arachnoid (Ar), but a few layers appear to continue over the roots as the inner layer of the root sheath (RS). The arachnoid is reflected over the roots at the SA and becomes continuous with the external layers of the RS. At the junction with the spinal cord, the outer layers become continuous with the pia mater (PM). Immediately beyond the spinal ganglion (SG), at the SA, the ventral and dorsal nerve roots unite to form the spinal nerve, which emerges through the intervertebral foramen and divides into a dorsal ramus (DRSN) and a ventral ramus (VRSN)?. Therefore intrathecal nerve roots are covered by an elastic root sheath derived from the arachnoid, whereas spinal nerves possess epi-perineurium which is relatively inelastic. Proximal-to-distal early GBS inflammatory lesions are illustrated as follows: ventral lumbar root (level 1), spinal nerve (level 2) and sciatic nerve (level 3). At level 1, this semithin complete cross section of ventral L5 root shows preservation of the density myelinated fibers (1A), though inflammatory lesions, observable at higher augmentation (not shown), may account for increased surface area, and thickening and contrast enhancement of ventral roots on spinal MRI (1B, arrows). Both cartoons at level 2 illustrate the following features: i/ normal anatomy of spinal nerve, usually monofascicular with epi-perineurial covering (2A), which account for its sonographic appearance usually consisting of a hypoechoic oval structure surrounded by hyperechoic perineurial rim; and ii/ endoneurial inflammatory oedema may cause a critical elevation in EFP that constricts transperineurial vessels by stretching the perineurium beyond the compliance limits (2B, arrowheads), which could result in areas of endoneurial ischemia, here centrofascicular. As illustrated herein (2A vs 2B), despite low spinal nerve compliance, early inflammatory events in GBS may cause an increase of cross sectional area (for images, see references [6,11). At level 3, this cross semithin section of sciatic nerve from a fatal AIDP patient shows several myelinated fibers exhibiting wallerian-like degeneration (myelin collapse, arrows) secondary to more proximal inflammatory lesions; note the presence of remyelinated fibres (arrowheads) and lipid-laden macrophages. Without knowledge of proximal nerve pathology, such distal florid wallerian-like lesions would make it very difficult to reach an accurate diagnosis. Diagram inspired by figure 3-6 from Berthold and colleagues [19].
A Kind of Epilogue
Therefore, the notion of the relevance of proximal nerve trunk inflammatory edema in classic VEGBS has pathophysiological, diagnostic, and therapeutic implications8: i/ in itself explains severe muscle weakness in patients showing minimal or no changes on conventional nerve conduction studies; ii/ its detection may require special electrophysiological techniques or the use of imaging techniques including ultrasonography; iii/endoneurial ischemia of extrathecal nerve trunks could account for increased serum neurofilament light chain concentration in early stages of both AIDP and AMAN [22,23]; and iv/ given the narrow therapeutic window to avoid the impact of edema on axons, there is a pressing need to implement measures to inhibit edema. The potential use of intravenous methylprednisolone in classic, severe VEGBS (500 mg daily for 5 consecutive days) deserves a special mention [1].
Several randomized controlled studies studying the effects of immunotherapy in GBS have been done in the last decades [1,2]. Intravenous immunoglobulin and plasma exchange have proved effective in patients unable to walk unaided, especially when less than 2 weeks from the onset weakness. Contrariwise, in the meta-analysis of the two trials of intravenous methylprednisolone, there was a non-significant trend towards benefit from corticosteroids [24]. However, early EAN treatment with high-dose corticosteroids does suppress the clinical deficit and haste recovery. As aforementioned, in the first few days of severe classic GBS the predominant pathological change is endoneurial inflammatory edema in proximal nerve trunks; in those possessing epi-perineurium edema is not an innocent bystander, but rather a potential cause of nerve conduction failure, eventually followed by distally accentuated Wallerian-like degeneration. Under such circumstances, this is the rationale for adding, as soon as possible, pulses of intravenous methylprednisolone to conventional immunotherapy [25-27].
Conclusion
Inflammatory oedema of proximal nerve trunks is the outstanding VEGBS pathological event, a notion that has pathophysiological, diagnostic and therapeutic implications.
Acknowledgement
There was no funding provided for this paper.
Conflict of Interest
The author declares no conflict of interest.
References
1. van Doorn PA, Ruts L, Jacobs BC (2008) Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol 7: 939-950. https://doi.org/10.1016/S1474-4422(08)70215-1
2. Willison HJ, Jacobs BC, van Doorn PA (2016) Guillain-Barré syndrome. Lancet 388: 717-727. https://doi.org/10.1016/S0140-6736(16)00339-1
3. Wakerley BR, Uncini A, Yuki N, et al. (2016) Guillain-Barré and Miller Fisher syndromes-new diagnostic classification. Nat Rev Neurol 10: 537-44. https://doi.org/10.1038/nrneurol.2014.138
4. Soliven B (2014) Animal models of autoimmune neuropathy. ILARJ 54: 282-290. https://doi.org/10.1093/ilar/ilt054
5. Uncini A, Kuwabara S (2012) Electrodiagnostic criteria for Guillain-Barré syndrome: a critical revision and the need for an update. Clin Neurophysiol 123: 1487-1495. https://doi.org/10.1016/j.clinph.2012.01.025
6. Berciano J, Orizaola P, Gallardo E, et al. (2020) Very early Guillain-Barré syndrome: A clinical-electrophysiological and ultrasonographic study. Clin Neurophysiol Pract 5: 1-9. https://doi.org/10.1016/j.cnp.2019.11.003
7. Nedkova V, Gutiérrez-Gutiérrez G, Navacerrada-Barrero FJ, et al. (2021) Re-evaluating the accuracy of optimized electrodiagnostic criteria in very early Guillain-Barré syndrome: a sequential study. Acta Neurol Belg 121: 1141-1150. https://doi.org/10.1007/s13760-021-01603-7
8. Berciano J (2021) Axonal degeneration in Guillain-Barré syndrome: a reappraisal. J Neurol 268: 5128-5143. https://doi.org/10.1007/s00415-020-10034-y
9. Haymaker WE, Kernohan JW (1949) The Landry-Guillain-Barré syndrome; a clinicopathologic report of 50 fatal cases and a critique of the literature. Medicine (Baltimore) 28: 59-141.
10. Krücke W (1955) Die primär-entzündliche Polyneuritis unbekannter Ursache. In: Lubarsch O, et al. eds., Handbuch der speziallen pathologischen Anatomie und Histologie, Vol XIII/5, Erkrankungen des peripheren und des vegetativen Nerven. Berlin, Springer-Verlag.
11. Gallardo E, Sedano MJ, Orizaola P, et al. (2015) Spinal nerve involvement in early Guillain-Barré syndrome: a clinico-electrophysiological, ultrasonographic and pathological study. Clin Neurophysiol 126: 810-819. https://doi.org/10.1016/j.clinph.2014.06.051
12. McKhann GM, Cornblath DR, Griffin JW, et al. (1993) Acute motor axonal neuropathy: a frequent cause of acute flaccid paralysis in China. Ann Neurol 33: 333-342. https://doi.org/10.1002/ana.410330402
13. Izumo S, Linington C, Wekerle H, et al. (1985) Morphologic study on experimental allergic neuritis mediated by T cell line specific for bovine P2 protein in Lewis rats. Lab Invest 53: 209-218.
14. Powell HC, Myers RR, Mizisin AP, et al. (1991) Response of the axon and barrier endothelium to experimental allergic neuritis induced by autoreactive T cell lines. Acta Neuropathol 82: 364-377. https://doi.org/10.1007/bf00296547
15. Berciano J, García A, Figols J, et al. (2000) Perineurium contributes to axonal damage in acute inflammatory demyelinating polyneuropathy. Neurology 55: 552-559. https://doi.org/10.1212/wnl.55.4.552
16. Berciano J, García A, Villagrá NT, et al. (2009) Severe Guillain-Barré syndrome: sorting out the pathological hallmark in an electrophysiological axonal case. J Peripher Nerv Syst 14: 54-63. https://doi.org/10.1111/j.1529-8027.2009.00206.x
17. Incesu TK, Secil Y, Tokucoglu F, et al. (2013) Diagnostic value of lumbar root stimulation at the early stage of Guillain-Barré syndrome. Clin Neurophysiol 124: 197-203. https://doi.org/10.1016/j.clinph.2012.07.004
18. Sevy A, Grapperon AM, Campana ES, et al. (2018) Detection of proximal conduction blocks using a triple stimulation technique improves the early diagnosis of Guillain-Barré syndrome. Clin Neurophysiol 129: 127-32. https://doi.org/10.1016/j.clinph.2017.10.035
19. Berthold CH, Fraher JP, King RHM, et al. (2005) Microscopical anatomy of the peripheral nervous system. In: Dyck PJ and Thomas PK. eds., Peripheral neuropathy. Philadelphia, WB Saunders 1: 35-91.
20. Berciano J, Sedano MJ, Pelayo-Negro AL, et al. (2017) Proximal nerve lesions in early Guillain-Barré syndrome: implications for pathogenesis and disease classification. J Neurol 264: 221-236. https://doi.org/10.1007/s00415-016-8204-2
21. Berciano J, Figols J, García A, et al. (1997) Fulminant Guillain-Barré syndrome with universal inexcitability of peripheral nerves: a clinicopathological study. Muscle Nerve 20: 846-857. https://doi.org/10.1002/(SICI)1097-4598(199707)20:7<846::AID-MUS9>3.0.CO;2-7
22. Altmann P, De Simoni D, Kaider A, et al. (2020) Increased serum neurofilament light chain concentration indicates poor outcome in Guillain-Barré syndrome. J Neuroinflammation 17: 86. https://doi.org/10.1186/s12974-020-01737-0
23. Martín-Aguilar L, Camps-Renom P, Lleixà C, et al. (2020) Serum neurofilament light chain predicts long-term prognosis in Guillain-Barré syndrome patients. J Neurol Neurosurg Psychiatry 92: 4. https://doi.org/10.1136/jnnp-2020-323899
24. Hughes RA, Brassington R, Gunn AA, et al. (2016) Corticosteroids for Guillain-Barré syndrome. Cochrane Database Syst Rev 10: CD001446. https://doi.org/10.1002/14651858.cd001446.pub5
25. Powell HC, Myers RR (1996) The axon in Guillain-Barré syndrome: immune target or innocent bystander? Ann Neurol 39: 4-5. https://doi.org/10.1002/ana.410390103
26. Berciano J (2021) The rationale for the use of corticosteroids in early severe Guillain-Barré syndrome. Autoimmun Rev 20: 102907. https://doi.org/10.1016/j.autrev.2021.102907
27. Berciano J (2021) Pathogenic events in very early Guillain-Barré syndrome: neither demyelination nor axonal degeneration but endoneurial inflammatory oedema. J Neurol 268. https://doi.org/10.1007/s00415-021-10773-6