A Double Jeopardy: Loss of FMRP Results in DSB and Down-regulated DNA Repair
Authors: Arijita Chakraborty1,3, Andre Grageda1,2, Vladimir A. Kuznetsov1,2, and Wenyi Feng1*
1Department of Biochemistry and Molecular Biology, SUNY Upstate Medical University, Syracuse, New York, USA
2Department of Urology, SUNY Upstate Medical University, Syracuse, New York, USA
3Tessera Therapeutics, Somerville, Massachusetts, USA
*Correspondence to: Wenyi Feng, Department of Biochemistry and Molecular Biology, SUNY Upstate Medical University, 750 East Adams Street, Syracuse, New York 13210, USA; E-mail: fengw@upstate.edu
Received: 20 July 2022; Revised: 11 October 2022; Accepted: 12 October 2022; Published: 17 October 2022
ORCID
Arijita Chakraborty: 0000-0002-4616-4399
Andre Grageda: 0000-0001-7532-1499
Vladimir A. Kuznetsov: 0000-0003-3405-0762
Wenyi Feng: 0000-0002-6123-7998
Citation: Arijita Chakraborty, Andre Grageda, Vladimir A. Kuznetsov and Wenyi Feng (2022) A double jeopardy: Loss of FMRP results in DSB and down-regulated DNA repair, 21st Century Pathology, Volume 2 (5): 125
Abstract
Our understanding of the molecular functions of the nucleocytoplasmic FMRP protein, which, if absent or dysfunctional, causes the Fragile X syndrome (FXS), largely revolves around its involvement in protein translation regulation in the cytoplasm. Recent studies have begun honing in on the nuclear and genomic functions of FMRP. We have shown that during DNA replication stress cells derived from FXS patients sustain increased level of R-loop formation and DNA double strand breaks. Here, we describe a transcriptomic analysis of these cells in order to identify those genes most impacted by the loss of FMRP with and without replication stress. We show that FMRP loss causes transcriptomic changes previously reported in untreated conditions. Importantly, we also show that replication stress, in addition to causing excess of DSB, results in down-regulation of transcription in virtually all DNA repair pathways. This finding suggests that despite normal DNA damage response, FXS patient-derived cells experience R-loop-induced DNA breakage as well as impaired DNA repair functions, effectively a double jeopardy. We suggest that it is imperative to deepen the understanding of the nuclear functions, particularly a genome protective function, of FMRP, which will lead to discoveries of novel therapeutic interventions for the FXS.
Keywords:
DNA double strand breaks (DSBs); DNA repair; FMR1; FMRP; Fragile X Syndrome (FXS); Genome instability; R-loop
Authors’ Contribution:
Conceptualization: AC & WF. Data curation: AC. Formal analysis: AC, AG & WF. Funding acquisition: VAK & WF.
Methodology: AC & WF. Project administration: WF. Visualization: AC & AG. Writing - original draft: AC. Writing - review & editing: AC & WF.
Conflict of Interest
No potential conflict of interest relevant to this article was reported.
Introduction
Fragile X syndrome (FXS) is a neurodevelopmental disorder affecting 1 in 7000 males and 1 in 11,000 females (National Fragile X foundation). FXS is the most common cause for inherited intellectual disability and developmental delay [1]. At the molecular level, FXS individuals-derived cells when cultured in folate deficient medium present a secondary constriction in the long arm of the X chromosome [2,3]. This abnormality defined the first rare fragile site associated with a genetic disorder, FRAXA (Figure 1A) [4]. It also constitutes the most frequent monogenic cause for autism spectrum disorder [5,6]. FXS patients display a multitude of behavioral problems such as anxiety, aggression, and attention deficit hyperactivity disorder [7]. FXS patients have limited treatment options with no cure and life-long dependency on psychopharmacological drugs to manage the behavioral problems [8].
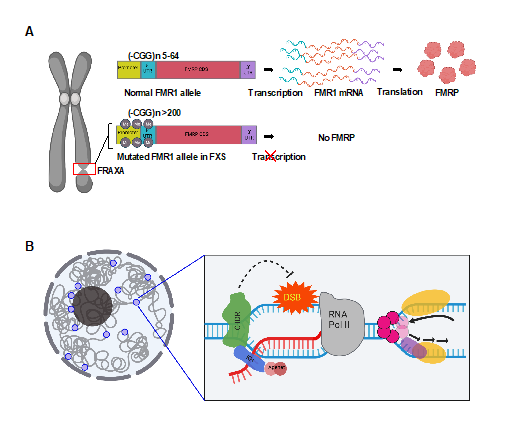
Figure 1: Overview of our current understanding of FXS. (A) A constriction in the long arm of the X chromosome marked by a red box represents the FRAXA site which is recurrently observed in FX cells under folate deficiency (left). The same site bears the mutated FMR1 gene. The 5’-UTR of the FMR1 gene has greater than 200 repeats for a full mutation. (B) A proposed genome protective role of FMRP as a novel R-loop regulator. FMRP inhibits R-loop mediated replication-transcription collision. FMRP interacts with the chromatin, binds R-loop directly and engages R-loop resolvases to initiate resolution thereby preventing DSBs. Images were created with BioRender.com.
FXS is primarily caused by CGG repeat expansion in the 5’UTR of the FMR1 gene, resulting in epigenetic silencing and lack of FMRP expression [9-12]. It is also less frequently caused by mutations in the coding region of FMR1 and thus dysfunctional FMRP [13,14]. FMRP is an RNA-binding protein and has multifaceted functions. It regulates key neuronal pathways by sequestering specific mRNA substrates and controlling signaling cascades across several cellular membrane receptors such as the metabotropic-glutamate receptor (mGluR), AMPA, NMDA, dopamine and cannabinoid receptors [15]. FMRP loss affects dendrite morphogenesis, neuronal circuit integration and axon guidance [16]. FMRP also interacts with pre-synaptic ion channels in hippocampal and cortical excitatory neurons and modulate neurotransmitter release and synaptic transmission [17-21]. Among FMRP's multi-faceted functions, the best understood is the mGluR-mediated long-term depression (LTD) pathway in which FMRP functions as a translation repressor [22]. Loss of FMRP causes an exaggerated mGluR-LTD and reduced synaptic strength [22-25]. However, despite the rescue of AMPA receptor trafficking defects in cultured neurons and behavior phenotypes in animal models, mGluR antagonists did not show expected efficacy in clinical trials [24,26-28]. Importantly, only a few of the mRNA targets of FMRP show high levels of protein expression in its absence and increased protein levels does not correlate with pathogenicity [29]. Therefore, it stands to reason that FMRP may have translation regulation-independent functions which underlie FXS disease etiology.
Since the discovery of FMRP as an mRNA binding protein, there has been an explosion of studies aiming to determine cell type- and sequence-specific binding of the mRNA targets of FMRP. The initial studies applied FXS mouse models with isolated brain regions (forebrains, hippocampus, cortex, cerebellum), followed by Purkinje cells and CA1 neurons, using RNA pull-down assays coupled with microarray or high-throughput sequencing [23,30-34]. Among these studies, it was reported that the FMRP mRNA targets were enriched in G-quadruplex sequences and/or long coding sequences and 3’UTRs. However, these studies in the mouse model do not correlate well with those using human counterparts in the majority of brain development [35]. Therefore, studies investigating FMRP mRNA targets in HEK293 cells and in adult post-mortem brain were conducted [36, 37], which led to the report of approximately 6000 human mRNA targets of FMRP [36]. A more recent study used human induced pluripotent stem cells differentiated into dorsal and ventral forebrain neural progenitor cells, arguably the most relevant cell types affected in FXS [38]. It showed that the FMRP tends to bind coding sequences instead of 3’UTRs, contrary to the mouse model, and preferably in long genes. Altogether, these studies did not reach an agreement on the mRNA sequence motifs that FMRP recognizes, suggesting that the recognition is structure- rather than sequence-specific, and is determined by the cell type. Importantly, genes whose mRNAs are FMRP binding targets participate in pathways that involve synaptic development, cell signaling, RNA transport, actin cytoskeleton, transcription, and epigenetic function [16,39]. Additionally, these genes are implicated in autism, thereby associating their binding by FMRP to potential disease mechanisms [23,37]. But what steps during mRNA regulatory or metabolic pathways other than translation regulation in which does FMRP function?
Studies in various model systems have now shown that FMRP functions in pre-mRNA splicing [40], mRNA stability [29,41], mRNA editing [42,43], and miRNA regulation [44,45]. In addition, studies have described nuclear and genomic functions of FMRP in DNA damage response, etc., which are not well understood [46-50]. We recently reported that lymphoblastoid cells derived from an FXS patient (FX cells) sustained genome-wide DNA double-strand breaks (DSBs) when undergoing DNA replication stress by aphidicolin (APH, a DNA polymerase inhibitor) [51]. Moreover, DSBs occurred near sequences that are prone to forming DNA:RNA hybrids called R-loops during gene transcription [51]. We also demonstrated that these FX cells have an intact DNA damage response [51]. These findings suggested a new co-transcriptional function of FMRP, which mitigates R-loop-induced DSBs during replication stress, thereby maintaining genome stability (Figure 1B). To further investigate this function, we asked if and how FMRP loss impacts the transcriptome upon replication stress in the FX lymphoblastoids in which we have analyzed DSB formation. Transcriptomic studies have primarily been conducted using brain tissue or cells from animal models of FXS. Due to cell heterogeneity, these studies have reported only subtle changes in mRNA levels [29,41,52], though single cell transcriptomics revealed dysregulation of cellular and molecular networks in the mouse model of FXS [41]. In humans, access to brain tissue is limited to adult post-mortem brain which does not model the neurodevelopmental role of FMRP. We note that peripheral blood cells have been used for molecular and phenotypic analyses of the FXS, as well as other autism spectrum disorders [1,53-55]. Our studies thus far have demonstrated that they are also a useful system for studying the genomic functions of FMRP.
Method
RNA-seq: FX cells and normal control (NM) cells were either treated with DMSO, 0.3 πM APH or left untreated for 24 h before harvest. 3x106 cells were harvested for RNA-seq. RNA was extracted using the Qiagen RNeasy Plus Mini Kit. The RNA was run on an Agilent 2100 Bioanalyzer using the RNA 6000 Nano Chip to assess RNA quality and quantity. 1 μg of total RNA was used as input to the Illumina TruSeq Stranded Total RNA Library Prep Kit Ribo Zero Gold H/M/R. Library size was assessed using the DNA 1000 chip on the Bioanalyzer, and the libraries were quantified using a Qubit fluorometer. Pair-end sequencing was run on an Illumina NextSeq 500 instrument. A total of four replicates were processed for treatment/conditions out of which three were biological replicates.
RNA-seq data analysis: Raw reads were obtained from Illumina Base space and pair-end reads were merged. Merged sequence reads were then aligned to the UCSC human genome assembly, GRCh37/hg19 using STAR-fusion aligner. The BAM files generated by STARfusion were then subjected to featureCounts [56] for the generation of read counts per gene. RNA-seq expression count obtained from featureCounts was Log2 transformed, mean normalized, and value trimmed prior to differential gene expression analysis. Mean normalization was performed by calculating the mean expression of every given sample. The mean of the sample means for each unique cell type and condition was then calculated. A correction coefficient was calculated by dividing a sample’s gene expression mean by their cell type and condition’s mean. Each sample was then multiplied by this correction coefficient. A cut-off value of 2(2^2=4 for raw counts) was used to determine genes which are not expressed as compared to genes that are expressed. Fold change was calculated by subtracting Log2 mean expression values and then setting 2 to the power of this value. Significance was determined by one-way ANOVA. The Benjamini & Hochberg method was used to calculate false discovery rate (FDR). Significant differentially expressed genes (DEGs) are determined by a p-value <= 0.05. Up-regulated and down-regulation of genes is determined by have a fold-change of >1 and <1 respectively.
Gene ontology analysis: Pathway analysis was performed using Enrichr [57,58]. Tables were generated using all significant DEGs, as well as significant up and down regulated DEGs. Databases used for this analysis include GO Molecular Function 2018, GO Cellular Component 2018, GO Biological Process 2018, WikiPathways 2019 Human, KEGG 2019 Human, Reactome 2016, InterPro Domains 2019, and Panther 2016. Pathways analysis was also performed on FDR significant (FDR <= 0.05) genes for each pair. Heatmaps were produced using Morpheus (https://software.broadinstitute.org/morpheus).
Results
We conducted a transcriptome analysis using total RNA isolated from FX and normal control (NM) lymphoblastoids, with and without replication stress by APH. We aimed to comprehensively define the transcriptomic changes due to FMRP loss and to address the increased DNA DSB phenotype in our previous studies. We performed differential gene expression analysis, comparing transcript counts in FX over NM cells. We categorized genes based on their transcriptional status (“on” vs. “off”) or expression level (“up-regulated” vs. “down-regulated”) in FX cells with respect to NM cells. Specifically, “on” corresponds to gene expression only in FX cells and not in NM cells, and vice versa for “off” genes. Similarly, “up- or down-regulated” correspond to genes expressed in both cell lines and with increased or decreased expression in FX cells compared to NM cells, respectively. First, there were more “on” than “off” genes in all conditions, suggesting a significant increase of transcriptional induction due to the loss of FMRP. Second, there were more up-regulated than down-regulated genes in both untreated and DMSO-treated conditions; however, the APH treatment caused a sharp increase of down-regulated genes by approximately 3-fold (Figure 2A). These results together suggest that, despite increasing transcriptional induction (without APH) in FX cells, replication stress by APH reduced the levels of gene expression. This is consistent with the notion that DNA damage itself has a negative impact on gene transcription [59] and that FX cells sustain higher level of DNA damage.
We next asked what biological pathways were enriched in the differentially expressed genes. In all conditions, genes up-regulated in cancer, such as “interferon alpha/beta signaling”, were up-regulated in FX cells (Figure 2B). For example, IFITM3 (Interferon-inducible Transmembrane Protein 3) has been recently associated with bone metastasis of prostate cancer cells [60]. Currently it is unclear if and how the up-regulated pathways impact FXS pathology, as cohort studies have reported conflicting conclusions as to whether FXS patients have increased risk for cancer [61,62]. However, we note that antiepileptic drug use, which is a common medical intervention among FXS patients, has been linked to increased risk for cancer [63]. Thus, it is challenging to delineate the cause for the observed up-regulation of cancer genes in FX cells. Additional up-regulated pathways include ‘immune response’, ‘Cytokine signaling’ and ‘Actin cytoskeleton regulation’, as reported by previous transcriptome studies [64,65]. On the other hand, genes involved in translation, including “eukaryotic translation elongation”, “3’-UTR-mediated translational regulation”, “major pathway of rRNA processing in the nucleolus” and “ribosome biogenesis”, were down-regulated in FX cells, presumably as a response to increased translational burden in the absence of FMRP. In contrast, APH caused down-regulation of 101 DNA repair genes and 29 G2/M checkpoint genes in FX cells (Figure 2B&C). This observation recapitulated a previous studies reporting down-regulated expression of DNA damage/repair pathway transcripts in FXS patient lymphoblastoids even without replication stress [55,65]. These results suggest that FX cells are inflicted with a double jeopardy during replication stress-that is-increased R-loop/DSB formation and down-regulated DNA repair.
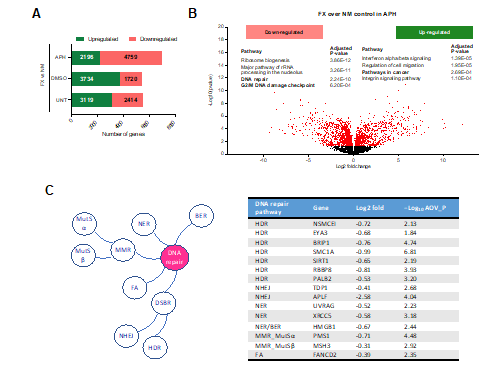
Figure 2: Emerging molecular players in FXS pathology identified by ChIP-seq and RNA-seq analysis. NM and FX cells were treated with DMSO, 0.3 μM APH or nothing for 24 h before harvest. 3x106 cells were used for RNA-seq using the Illumina TruSeq Stranded Total RNA Library Prep Kit Ribo Zero Gold H/M/R, with pair-end sequencing on Illumina NextSeq 500. Four replicates were processed. Detailed RNA-seq data analysis and raw data are accessible from the GEO accession number GSE124403. (A&B) Summary of gene expression from RNA-seq analysis. (A) Number of genes up- or down-regulated in FX cells when compared to NM cells with or without APH. (B) Volcano plot of -Log10 (p-value for significance in differential expression) versus Log2 (fold change of transcript levels of FX_APH to NM_APH) for all genes. Relative to NM_APH, significantly different genes in FX_APH with -Log10 p-value greater than 1.3 are shown in red. Top biological pathways that are enriched for those genes significantly down- or up-regulated in FX_APH cells relative to NM_APH are shown. (C) Representative down-regulated DNA repair genes in FX_APH cells. Log2 (fold change of expression of FX to control). AOV_P, differential expression ANOVA test P value. HDR, homologous DNA recombination; NHEJ, non-homologous end joining; NER, nucleotide excision repair; BER, base excision repair, MMR, mismatch repair; FA, Fanconi anemia pathway.
Lastly, we performed an integrative analysis of DSBs and gene expression in FX cells. We have previously observed that 2673 genes suffered DSBs specifically in APH-treated FX cells. Indeed, 142 of the 2673 genes were differentially expressed and 86 were down-regulated in APH-treated FX cells. Interestingly, these genes were enriched in the RNA Pol II transcription pathway and specifically, transcription regulation by p53, including BRCA1, CARM1, FANCD2, FANCI, RABGGTB, RPTOR, USP7, YWHAE, and ZNF420 (Figure 3), further strengthening the down-regulation of DNA repair. These observations also suggested that DSBs in genes that code for transcription factors may have contributed to the down-regulation of DNA repair genes, in addition to the lack of direct protection by FMRP.
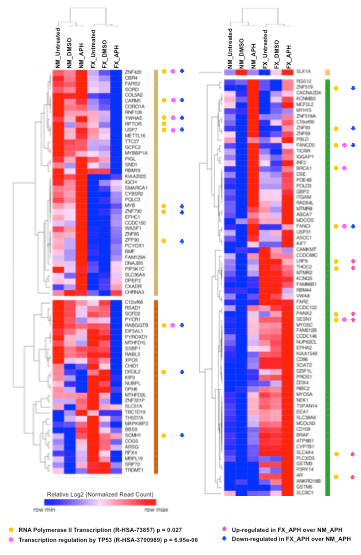
Figure 3: TP53-regulated transcription pathway genes are down-regulated in FX cells undergoing APH-induced DSBs. Heat map analysis and hierarchical clustering of gene expression from 142 protein coding genes suffering DSBs in FX cells specifically under APH treatment [51]. Log2 transformed normalized read counts were used to perform the analysis using Morpheus (https://software.broadinstitute.org/morpheus). Reactome pathways enriched for differentially expressed genes in APH-treated FX cells (FX_APH) relative to the NM_APH cells are indicated with solid circles. Up- and down-regulated genes are indicated by up and down arrows, respectively.
Discussion
Our previous study led us to conclude that the FX genome suffers from R-loop-associated DSBs induced by replication stress [51]. Among the DSB hotspots are many genes involved in neuronal development and synaptic regulation, suggesting that these genes are protected by FMRP in addition to being translationally regulated by it [51]. Thus, it appears that FMRP controls all aspects of RNA metabolism including co-transcriptional regulation. In this study we further demonstrated that APH-treated FX cells show down-regulated expression of genes in virtually all DNA repair pathways. This is a result that recapitulated previous findings of the FX cells without replication stress, though only a selected few DNA repair pathways were affected [55,65]. In addition, it has been shown that mouse embryonic fibroblasts from an FXS mouse model showed defective single-stranded DNA repair during meiotic DSB formation [46].
In contrast to the previous gene expression studies which used microarray-based gene expression data, we found that there were more up-regulated genes in FX cells compared to the control cells, suggesting that FX cells have heightened transcriptional response as a result of FMRP deficiency. The up-regulated genes are enriched in oncogenic pathways such as ‘Ras protein signaling transduction’ including MAPKAPK3, RAB genes, TIAM1, INFα/β and KRAS. Moreover, MDM2 and XIAP, which prevents p53 accumulation and inhibits apoptosis, respectively, are also up-regulated in our current study. Consistent with our finding, recent RNA-seq studies using neuronal cells differentiated from human embryonic stem cells or induced pluripotent stem cell models of FXS also reported up-regulated expression of PI3K-AKT and ERK/MAPK pathways, both of which are downstream to RAS signaling and controlled by the RAS proteins with implications of cancer-like transformations [64,66-69]. Interestingly, we also observed an increased expression of Amyloid β-precursor protein (APP) in FX cells compared to control without replication stress (Log2 fold change values 0.44 and 0.50 for untreated and DMSO-treated, respectively). Upon APH treatment the differential expression dropped to Log 2 fold change of 0.14. APP is an integral membrane protein that is ubiquitously expressed but enriched in the brain [70]. APP undergoes proteolytic cleavage by three types of proteases that results in the shedding of the extracellular domain. The type of proteolytic processing can result in neuroprotective or neurotoxic consequences as observed in Alzheimer’s disease with the accumulation of Aβ-peptide [71]. FMRP has been shown to bind APP mRNA directly, and through the miRNA pathway suppress its translation [71,72]. Consequently, APP and its cleavage products were found to be up-regulated in Fmr1 KO mice. Moreover, APP haploinsufficiency resulted in the rescue of repetitive behavior, hyperactivity, mGluR-LTD and spine morphology in a mice model of FXS [72]. Similarly, APP, sAPPα and Aβ peptides are shown to be up-regulated in post-mortem brain and in the blood plasma of FXS children [71,72]. Our findings suggest that the APP mRNA is regulated by FMRP both transcriptionally and translationally, in the absence of replication stress.
Treatment of the FX-patient derived cells with APH resulted in a shift in the mRNA expression pattern such that more genes were down-regulated because of DNA damage. Notably, we observed down-regulation of genes in virtually all DNA repair pathways.
Conclusion
In conclusion, our results suggest that the FX genome undergoes a double jeopardy of sustaining R-loop-induced DSBs and reduced DNA repair as a result of replication stress. APH treatment led to more genes showing down-regulated expression compared to vehicle control cells, possibly due to DNA damage of these genes. Indeed, 60% of the DEGs that also sustained DSBs in APH-treated cells showed decreased expression in APH. We envision that such genome instability may profoundly impact cellular functions of neuronal cells when FMRP is absent. It has not escaped our attention that post-mitotic neurons are unlikely subjected to DNA replication stress. However, we note that R-loop formation can be induced by chemicals/reagents that perturb gene transcription, thus still necessitating FMRP to resolve R-loops and maintain genome integrity. Future work would be dedicated to understanding of the mechanisms of FMRP protection of the mRNA substrates, particularly DNA repair genes, during transcription. It will also be dedicated to the determination of neuronal activities upon the loss and gain of FMRP’s genomic substrates that have been identified in the lymphoblastoid cells. In turn, this effort would likely lead to better targets for therapeutic interventions of FXS.
References
1. Ciaccio C, Fontana L, Milani D, Tabano S, Miozzo M, Esposito S. Fragile X syndrome: a review of clinical and molecular diagnoses. Italian journal of pediatrics. 2017 Dec;43(1):1-2. https://doi.org/10.1186/s13052-017-0355-y
2. Harvey J, Judge C, Wiener S. Familial X-linked mental retardation with an X chromosome abnormality. Journal of medical genetics. 1977 Feb 1;14(1):46-50. https://doi.org/10.1136/jmg.14.1.46
3. Phalen JA. Fragile X syndrome. Pediatr Rev. 2005;26:181-182. https://doi.org/10.1542/pir.26-5-181
4. Feng W, Chakraborty A. Fragility extraordinaire: unsolved mysteries of chromosome fragile sites. DNA Replication. 2017:489-526. https://doi.org/10.1007/978-981-10-6955-0_21
5. Bagni C, Zukin RS. A synaptic perspective of fragile X syndrome and autism spectrum disorders. Neuron. 2019 Mar 20;101(6):1070-88. https://doi.org/10.1016/j.neuron.2019.02.041
6. Lai A, Valdez?Sinon AN, Bassell GJ. Regulation of RNA granules by FMRP and implications for neurological diseases. Traffic. 2020 Jul;21(7):454-62. https://doi.org/10.1111/tra.12733
7. Abbeduto L, Thurman AJ, McDuffie A, Klusek J, Feigles RT, Ted Brown W, Harvey DJ, Adayev T, LaFauci G, Dobkins C, Roberts JE. ASD comorbidity in fragile X syndrome: Symptom profile and predictors of symptom severity in adolescent and young adult males. Journal of Autism and Developmental Disorders. 2019 Mar;49(3):960-77. https://doi.org/10.1007/s10803-018-3796-2
8. Davidson M, Sebastian SA, Benitez Y, Desai S, Quinonez J, Ruxmohan S, Stein JD, Cueva W. Behavioral Problems in Fragile X Syndrome: A Review of Clinical Management. Cureus. 2022 Feb 2;14(2). https://doi.org/10.7759/cureus.21840
9. Heitz D, Rousseau F, Devys D, Saccone S, Abderrahim H, Le Paslier D, Cohen D, Vincent A, Toniolo D, Della Valle G, Johnson S. Isolation of sequences that span the fragile X and identification of a fragile X-related CpG island. Science. 1991 Mar 8;251(4998):1236-9. https://doi.org/10.1126/science.2006411
10. Kremer EJ, Pritchard M, Lynch M, Yu S, Holman K, Baker E, Warren ST, Schlessinger D, Sutherland GR, Richards RI. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p (CCG) n. Science. 1991 Jun 21;252(5013):1711-4. https://doi.org/10.1126/science.1675488
11. Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang F, Eussen BE. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991 May 31;65(5):905-14. https://doi.org/10.1016/0092-8674(91)90397-h
12. Vincent A, Hertz D, Petit C, Kretz C, Oberlé I, Mandel JL. Abnormal pattern detected in fragile-X patients by pulsed-field gel electrophoresis. Nature. 1991 Feb;349(6310):624-6. https://doi.org/10.1038/349624a0
13. Sitzmann AF, Hagelstrom RT, Tassone F, Hagerman RJ, Butler MG. Rare FMR1 gene mutations causing fragile X syndrome: a review. American Journal of Medical Genetics Part A. 2018 Jan;176(1):11-8. https://doi.org/10.1002/ajmg.a.38504
14. Suhl JA, Warren ST. Single-nucleotide mutations in FMR1 reveal novel functions and regulatory mechanisms of the fragile X syndrome protein FMRP. Journal of experimental neuroscience. 2015 Jan;9:JEN-S25524. https://doi.org/10.4137/jen.s25524
15. Fernández E, Rajan N, Bagni C. The FMRP regulon: from targets to disease convergence. Frontiers in neuroscience. 2013 Oct 24;7:191. https://doi.org/10.3389/fnins.2013.00191
16. Richter JD, Zhao X. The molecular biology of FMRP: new insights into fragile X syndrome. Nature Reviews Neuroscience. 2021 Apr;22(4):209-22. https://doi.org/10.1038/s41583-021-00432-0
17. Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, Navaratnam D, Kaczmarek LK. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nature neuroscience. 2010 Jul;13(7):819-21. https://doi.org/10.1038/nn.2563
18. Deng PY, Rotman Z, Blundon JA, Cho Y, Cui J, Cavalli V, Zakharenko SS, Klyachko VA. FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron. 2013 Feb 20;77(4):696-711. https://doi.org/10.1016/j.neuron.2012.12.018
19. Ferron L, Nieto-Rostro M, Cassidy JS, Dolphin AC. Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nature communications. 2014 Apr 7;5(1):1-4. https://doi.org/10.1038/ncomms4628
20. Kshatri A, Cerrada A, Gimeno R, Bartolomé-Martín D, Rojas P, Giraldez T. Differential regulation of BK channels by fragile X mental retardation protein. Journal of General Physiology. 2020 Jun 1;152(6). https://doi.org/10.1085/jgp.201912502
21. Yang YM, Arsenault J, Bah A, Krzeminski M, Fekete A, Chao OY, Pacey LK, Wang A, Forman-Kay J, Hampson DR, Wang LY. Identification of a molecular locus for normalizing dysregulated GABA release from interneurons in the Fragile X brain. Molecular psychiatry. 2020 Sep;25(9):2017-35. https://doi.org/10.1038/s41380-018-0240-0
22. Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends in neurosciences. 2004 Jul 1;27(7):370-7. https://doi.org/10.1016/j.tins.2004.04.009
23. Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011 Jul 22;146(2):247-61. https://doi.org/10.1016/j.cell.2011.06.013
24. Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proceedings of the National Academy of Sciences. 2007 Sep 25;104(39):15537-42. https://doi.org/10.1073/pnas.0707484104
25. Niere F, Wilkerson JR, Huber KM. Evidence for a fragile X mental retardation protein-mediated translational switch in metabotropic glutamate receptor-triggered Arc translation and long-term depression. Journal of Neuroscience. 2012 Apr 25;32(17):5924-36. https://doi.org/10.1523/jneurosci.4650-11.2012
26. Berry-Kravis E, Des Portes V, Hagerman R, Jacquemont S, Charles P, Visootsak J, Brinkman M, Rerat K, Koumaras B, Zhu L, Barth GM. Mavoglurant in fragile X syndrome: Results of two randomized, double-blind, placebo-controlled trials. Science translational medicine. 2016 Jan 13;8(321):321ra5-. https://doi.org/10.1126/scitranslmed.aab4109
27. McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, McDonald TV. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005 Mar 3;45(5):753-64. https://doi.org/10.1016/j.neuron.2005.01.038
28. Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005 Dec 1;49(7):1053-66. https://doi.org/10.1016/j.neuropharm.2005.06.004
29. Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang W, Song H, Wu H, Shu Q, Jin P. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Human molecular genetics. 2018 Nov 15;27(22):3936-50. https://doi.org/10.1093/hmg/ddy292
30. Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001 Nov 16;107(4):477-87. https://doi.org/10.1016/s0092-8674(01)00568-2
31. Maurin T, Lebrigand K, Castagnola S, Paquet A, Jarjat M, Popa A, Grossi M, Rage F, Bardoni B. HITS-CLIP in various brain areas reveals new targets and new modalities of RNA binding by fragile X mental retardation protein. Nucleic acids research. 2018 Jul 6;46(12):6344-55. https://doi.org/10.1093/nar/gky267
32. Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, Eberwine J. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003 Feb 6;37(3):417-31. https://doi.org/10.1016/s0896-6273(03)00034-5
33. Sawicka K, Hale CR, Park CY, Fak JJ, Gresack JE, Van Driesche SJ, Kang JJ, Darnell JC, Darnell RB. FMRP has a cell-type-specific role in CA1 pyramidal neurons to regulate autism-related transcripts and circadian memory. Elife. 2019 Dec 20;8:e46919. https://doi.org/10.7554/elife.46919
34. Van Driesche SJ, Sawicka K, Zhang C, Hung SK, Park CY, Fak JJ, Yang C, Darnell RB, Darnell JC. FMRP binding to a ranked subset of long genes is revealed by coupled CLIP and TRAP in specific neuronal cell types. BioRxiv. 2019 Jan 1:762500. https://doi.org/10.1101/762500
35. Kwan KY, Lam MM, Johnson MB, Dube U, Shim S, Rašin MR, Sousa AM, Fertuzinhos S, Chen JG, Arellano JI, Chan DW. Species-dependent posttranscriptional regulation of NOS1 by FMRP in the developing cerebral cortex. Cell. 2012 May 11;149(4):899-911. https://doi.org/10.1016/j.cell.2012.02.060
36. Ascano M, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, Williams Z. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012 Dec;492(7429):382-6. https://doi.org/10.1038/nature11737
37. Tran SS, Jun HI, Bahn JH, Azghadi A, Ramaswami G, Van Nostrand EL, Nguyen TB, Hsiao YH, Lee C, Pratt GA, Martínez-Cerdeño V. Widespread RNA editing dysregulation in brains from autistic individuals. Nature neuroscience. 2019 Jan;22(1):25-36. https://doi.org/10.1038/s41593-018-0287-x
38. Li M, Shin J, Risgaard RD, Parries MJ, Wang J, Chasman D, Liu S, Roy S, Bhattacharyya A, Zhao X. Identification of FMR1-regulated molecular networks in human neurodevelopment. Genome research. 2020 Mar 1;30(3):361-74. https://doi.org/10.1101/gr.251405.119
39. Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB, Moine H, Kooy RF, Tassone F, Gantois I, Sonenberg N, Mandel JL, Hagerman PJ. Fragile X syndrome. Nature reviews Disease primers. 2017 Sep 29;3(1):1-9. https://doi.org/10.1038/nrdp.2017.65
40. Zhou LT, Ye SH, Yang HX, Zhou YT, Zhao QH, Sun WW, Gao MM, Yi YH, Long YS. A novel role of fragile X mental retardation protein in pre-mRNA alternative splicing through RNA-binding protein 14. Neuroscience. 2017 May 4;349:64-75. https://doi.org/10.1016/j.neuroscience.2017.02.044
41. Shu H, Donnard E, Liu B, Jung S, Wang R, Richter JD. FMRP links optimal codons to mRNA stability in neurons. Proceedings of the National Academy of Sciences. 2020 Dec 1;117(48):30400-11. https://doi.org/10.1073/pnas.2009161117
42. Filippini A, Bonini D, Lacoux C, Pacini L, Zingariello M, Sancillo L, Bosisio D, Salvi V, Mingardi J, La Via L, Zalfa F. Absence of the Fragile X Mental Retardation Protein results in defects of RNA editing of neuronal mRNAs in mouse. RNA biology. 2017 Nov 2;14(11):1580-91. https://doi.org/10.1080/15476286.2017.1338232
43. Shamay-Ramot A, Khermesh K, Porath HT, Barak M, Pinto Y, Wachtel C, Zilberberg A, Lerer-Goldshtein T, Efroni S, Levanon EY, Appelbaum L. Fmrp interacts with adar and regulates RNA editing, synaptic density and locomotor activity in zebrafish. PLoS genetics. 2015 Dec 4;11(12):e1005702. https://doi.org/10.1371/journal.pgen.1005702
44. Golovin RM, Broadie K. Developmental experience-dependent plasticity in the first synapse of the Drosophila olfactory circuit. Journal of Neurophysiology. 2016 Dec 1;116(6):2730-8. https://doi.org/10.1152/jn.00616.2016
45. Sudhakaran IP, Hillebrand J, Dervan A, Das S, Holohan EE, Hülsmeier J, Sarov M, Parker R, VijayRaghavan K, Ramaswami M. FMRP and Ataxin-2 function together in long-term olfactory habituation and neuronal translational control. Proceedings of the National Academy of Sciences. 2014 Jan 7;111(1):E99-108. https://doi.org/10.1073/pnas.1309543111
46. Alpatov R, Lesch BJ, Nakamoto-Kinoshita M, Blanco A, Chen S, Stützer A, Armache KJ, Simon MD, Xu C, Ali M, Murn J. A chromatin-dependent role of the fragile X mental retardation protein FMRP in the DNA damage response. Cell. 2014 May 8;157(4):869-81. https://doi.org/10.1016/j.cell.2014.03.040
47. Dockendorff TC, Labrador M. The fragile X protein and genome function. Molecular neurobiology. 2019 Jan;56(1):711-21. https://doi.org/10.1007/s12035-018-1122-9
48. Fridell RA, Benson RE, Hua J, Bogerd HP, Cullen BR. A nuclear role for the Fragile X mental retardation protein. The EMBO journal. 1996 Oct;15(19):5408-14.
49. Liu W, Jiang F, Bi X, Zhang YQ. Drosophila FMRP participates in the DNA damage response by regulating G2/M cell cycle checkpoint and apoptosis. Human molecular genetics. 2012 Nov 1;21(21):4655-68. https://doi.org/10.1093/hmg/dds307
50. Zhang W, Cheng Y, Li Y, Chen Z, Jin P, Chen D. A feed-forward mechanism involving Drosophila fragile X mental retardation protein triggers a replication stress-induced DNA damage response. Human molecular genetics. 2014 Oct 1;23(19):5188-96. https://doi.org/10.1093/hmg/ddu241
51. Chakraborty A, Jenjaroenpun P, Li J, El Hilali S, McCulley A, Haarer B, Hoffman EA, Belak A, Thorland A, Hehnly H, Schildkraut CL. Replication stress induces global chromosome breakage in the fragile X genome. Cell reports. 2020 Sep 22;32(12):108179. https://doi.org/10.1016/j.celrep.2020.108179
52. Thomson SR, Seo SS, Barnes SA, Louros SR, Muscas M, Dando O, Kirby C, Wyllie DJ, Hardingham GE, Kind PC, Osterweil EK. Cell-type-specific translation profiling reveals a novel strategy for treating fragile X syndrome. Neuron. 2017 Aug 2;95(3):550-63. https://doi.org/10.1016/j.neuron.2017.07.013
53. Bittel DC, Kibiryeva N, Butler MG. Whole genome microarray analysis of gene expression in subjects with fragile X syndrome. Genetics in Medicine. 2007 Jul 1;9(7):464-72. https://doi.org/10.1097/gim.0b013e3180ca9a9a
54. Talebizadeh Z, Butler MG, Theodoro MF. Feasibility and relevance of examining lymphoblastoid cell lines to study role of microRNAs in autism. Autism Research. 2008 Aug;1(4):240-50. https://doi.org/10.1002/aur.33
55. Xu H, Rosales-Reynoso MA, Barros-Núñez P, Peprah E. DNA repair/replication transcripts are down regulated in patients with Fragile X Syndrome. BMC research notes. 2013 Dec;6(1):1-5. https://doi.org/10.1186/1756-0500-6-90
56. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014 Apr 1;30(7):923-30. https://doi.org/10.1093/bioinformatics/btt656
57. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC bioinformatics. 2013 Dec;14(1):1-4. https://doi.org/10.1186/1471-2105-14-128
58. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids research. 2016 Jul 8;44(W1):W90-7. https://doi.org/10.1093/nar/gkw377
59. Christmann M, Kaina B. Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic acids research. 2013 Jul 27;41(18):8403-20. https://doi.org/10.1093/nar/gkt635
60. Liu X, Chen L, Fan Y, Hong Y, Yang X, Li Y, Lu J, Lv J, Pan X, Qu F, Cui X. IFITM3 promotes bone metastasis of prostate cancer cells by mediating activation of the TGF-β signaling pathway. Cell death & disease. 2019 Jul 4;10(7):1-6. https://doi.org/10.1038/s41419-019-1750-7
61. Schultz?Pedersen S, Hasle H, Olsen JH, Friedrich U. Evidence of decreased risk of cancer in individuals with fragile X. American journal of medical genetics. 2001 Oct 15;103(3):226-30.
62. Sund R, Pukkala E, Patja K. Cancer incidence among persons with fragile X syndrome in Finland: a population?based study. Journal of Intellectual Disability Research. 2009 Jan;53(1):85-90. https://doi.org/10.1111/j.1365-2788.2008.01116.x
63. Lamminpää A, Pukkala E, Teppo L, Neuvonen PJ. Cancer incidence among patients using antiepileptic drugs: a long-term follow-up of 28,000 patients. European journal of clinical pharmacology. 2002 May;58(2):137-41. https://doi.org/10.1007/s00228-002-0429-6
64. Lu P, Chen X, Feng Y, Zeng Q, Jiang C, Zhu X, Fan G, Xue Z. Integrated transcriptome analysis of human iPS cells derived from a fragile X syndrome patient during neuronal differentiation. Science China Life sciences. 2016 Nov;59(11):1093-105. https://doi.org/10.1007/s11427-016-0194-6
65. Nishimura Y, Martin CL, Vazquez-Lopez A, Spence SJ, Alvarez-Retuerto AI, Sigman M, Steindler C, Pellegrini S, Schanen NC, Warren ST, Geschwind DH. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Human molecular genetics. 2007 Jul 15;16(14):1682-98. https://doi.org/10.1093/hmg/ddm116
66. Gimple RC, Wang X. RAS: Striking at the Core of the Oncogenic Circuitry. Frontiers in oncology. 2019 Sep 24;9:965. https://doi.org/10.3389/fonc.2019.00965
67. Haenfler JM, Skariah G, Rodriguez CM, Monteiro da Rocha A, Parent JM, Smith GD, Todd PK. Targeted reactivation of FMR1 transcription in fragile X syndrome embryonic stem cells. Frontiers in molecular neuroscience. 2018 Aug 15;11:282. https://doi.org/10.3389/fnmol.2018.00282
68. Kuznitsov-Yanovsky L, Shapira G, Gildin L, Shomron N, Ben-Yosef D. Transcriptomic Analysis of Human Fragile X Syndrome Neurons Reveals Neurite Outgrowth Modulation by the TGFβ/BMP Pathway. International journal of molecular sciences. 2022 Aug 17;23(16):9278. https://doi.org/10.3390/ijms23169278
69. Raj N, McEachin ZT, Harousseau W, Zhou Y, Zhang F, Merritt-Garza ME, Taliaferro JM, Kalinowska M, Marro SG, Hales CM, Berry-Kravis E. Cell-type-specific profiling of human cellular models of fragile X syndrome reveal PI3K-dependent defects in translation and neurogenesis. Cell reports. 2021 Apr 13;35(2):108991. https://doi.org/10.1016/j.celrep.2021.108991
70. Zheng H, Koo EH. Biology and pathophysiology of the amyloid precursor protein. Molecular neurodegeneration. 2011 Dec;6(1):1-6. https://doi.org/10.1186/1750-1326-6-27
71. Dionne O, Corbin F. An “omic” overview of fragile X syndrome. Biology. 2021 May 13;10(5):433. https://doi.org/10.3390/biology10050433
72. Westmark CJ. Fragile X and APP: a decade in review, a vision for the future. Molecular neurobiology. 2019 Jun;56(6):3904-21. https://doi.org/10.1007/s12035-018-1344-x